

Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits
- PMID: 24196873
- PMCID: PMC3875850
- DOI: 10.1101/gr.164079.113
Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits
Abstract
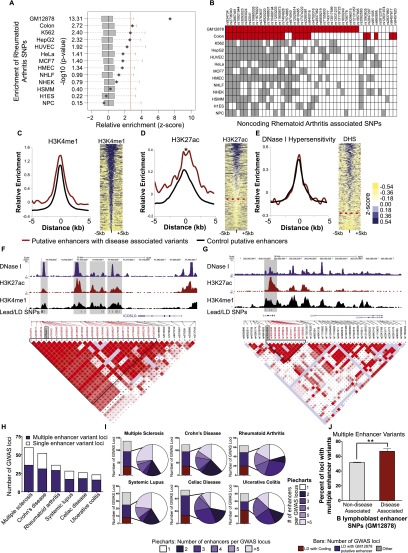
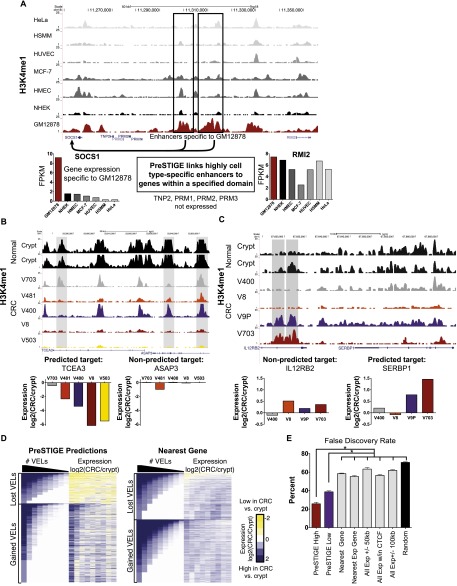
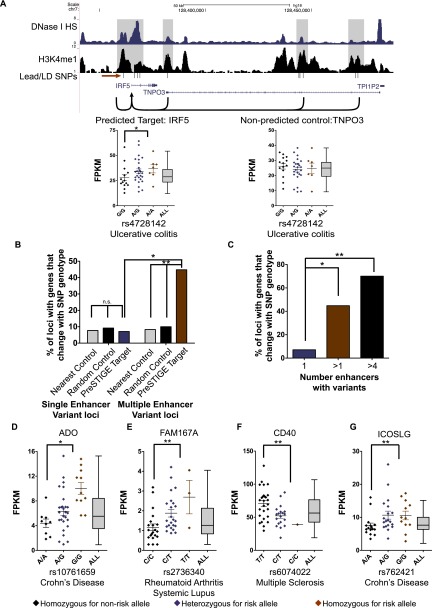
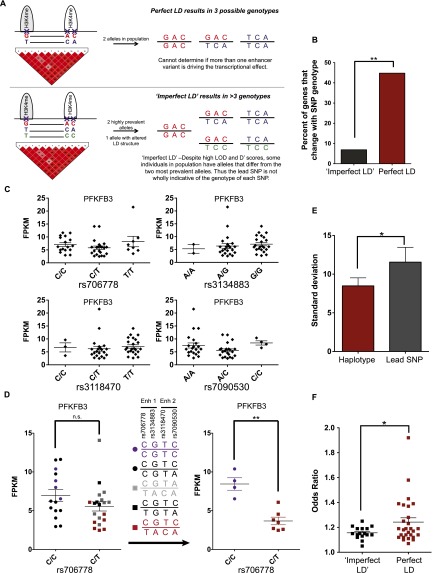
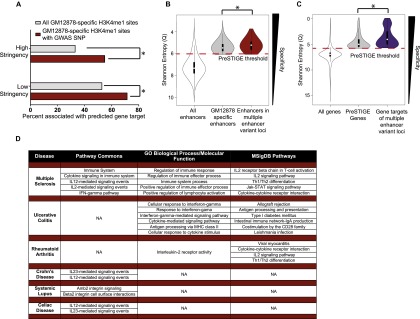
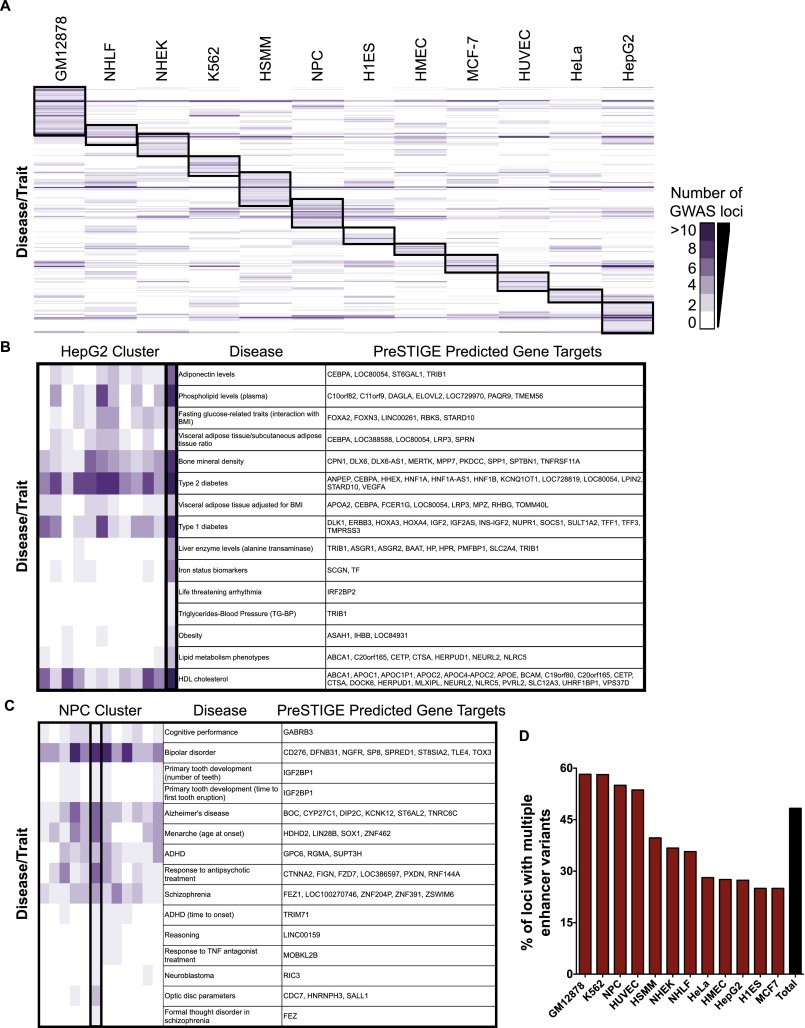
DNA variants (SNPs) that predispose to common traits often localize within noncoding regulatory elements such as enhancers. Moreover, loci identified by genome-wide association studies (GWAS) often contain multiple SNPs in linkage disequilibrium (LD), any of which may be causal. Thus, determining the effect of these multiple variant SNPs on target transcript levels has been a major challenge. Here, we provide evidence that for six common autoimmune disorders (rheumatoid arthritis, Crohn's disease, celiac disease, multiple sclerosis, lupus, and ulcerative colitis), the GWAS association arises from multiple polymorphisms in LD that map to clusters of enhancer elements active in the same cell type. This finding suggests a "multiple enhancer variant" hypothesis for common traits, where several variants in LD impact multiple enhancers and cooperatively affect gene expression. Using a novel method to delineate enhancer-gene interactions, we show that multiple enhancer variants within a given locus typically target the same gene. Using available data from HapMap and B lymphoblasts as a model system, we provide evidence at numerous loci that multiple enhancer variants cooperatively contribute to altered expression of their gene targets. The effects on target transcript levels tend to be modest and can be either gain- or loss-of-function. Additionally, the genes associated with multiple enhancer variants encode proteins that are often functionally related and enriched in common pathways. Overall, the multiple enhancer variant hypothesis offers a new paradigm by which noncoding variants can confer susceptibility to common traits.
Figures
References
-
- Barreiro LB, Quintana-Murci L 2010. From evolutionary genetics to human immunology: How selection shapes host defence genes. Nat Rev Genet 11: 17–30 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials