

Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation
- PMID: 24200680
- PMCID: PMC3901070
- DOI: 10.1182/blood-2013-10-530683
Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation
Abstract
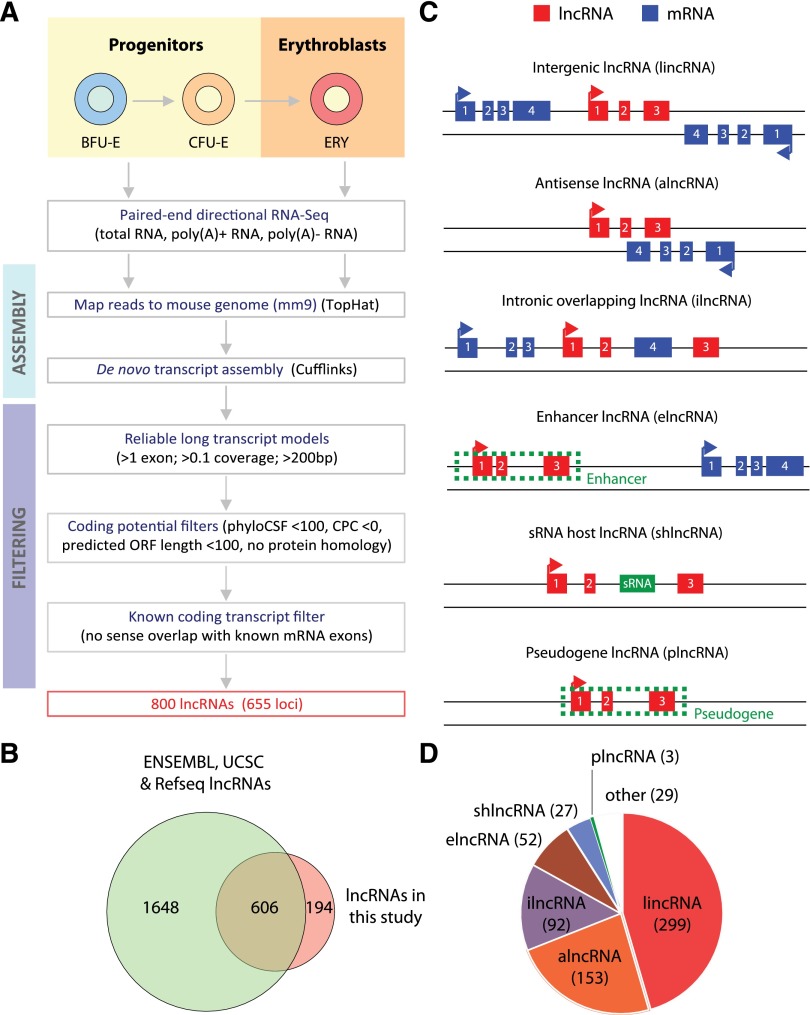
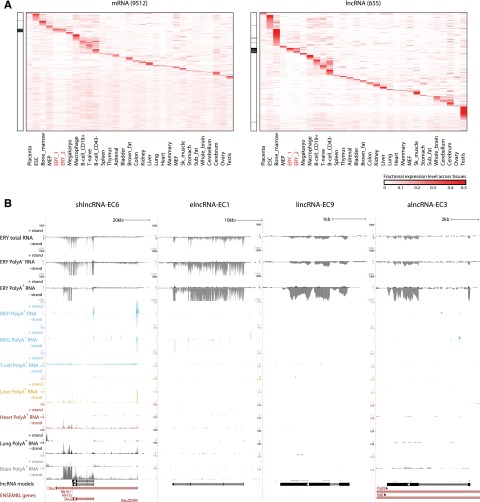
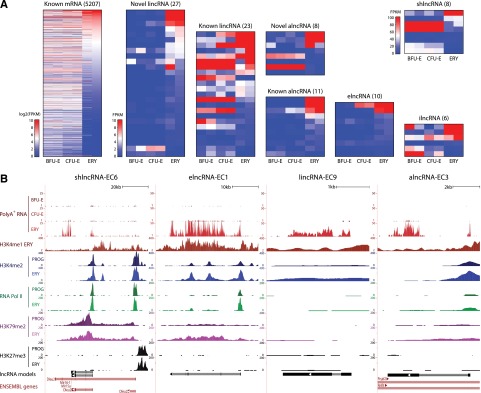
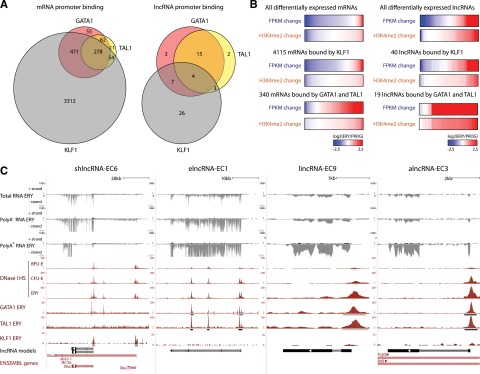
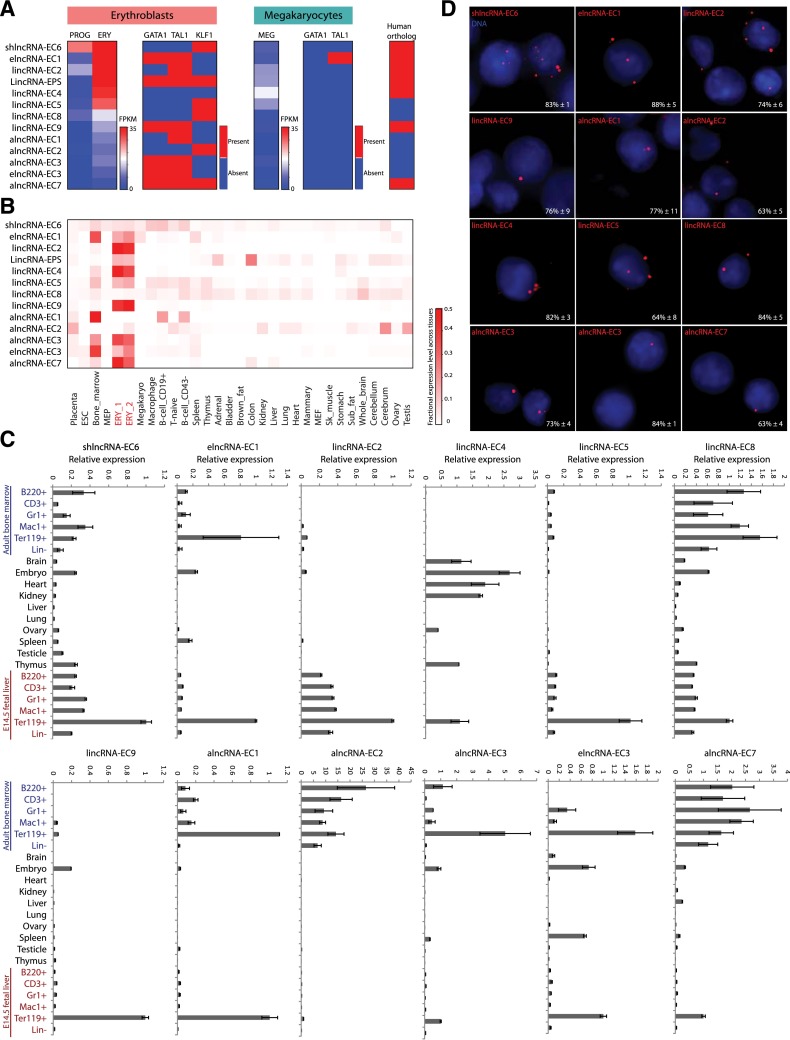
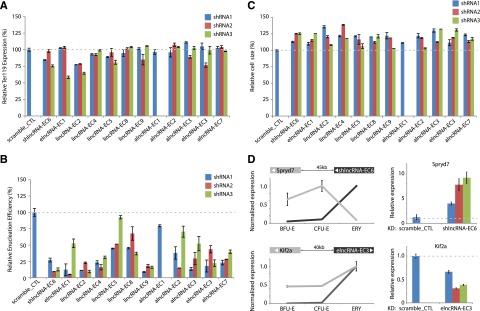
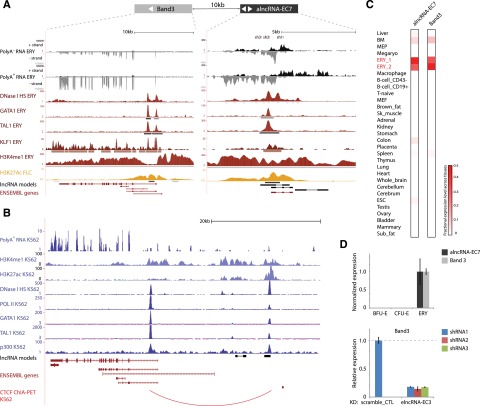
Erythropoiesis is regulated at multiple levels to ensure the proper generation of mature red cells under multiple physiological conditions. To probe the contribution of long noncoding RNAs (lncRNAs) to this process, we examined >1 billion RNA-seq reads of polyadenylated and nonpolyadenylated RNA from differentiating mouse fetal liver red blood cells and identified 655 lncRNA genes including not only intergenic, antisense, and intronic but also pseudogene and enhancer loci. More than 100 of these genes are previously unrecognized and highly erythroid specific. By integrating genome-wide surveys of chromatin states, transcription factor occupancy, and tissue expression patterns, we identify multiple lncRNAs that are dynamically expressed during erythropoiesis, show epigenetic regulation, and are targeted by key erythroid transcription factors GATA1, TAL1, or KLF1. We focus on 12 such candidates and find that they are nuclear-localized and exhibit complex developmental expression patterns. Depleting them severely impaired erythrocyte maturation, inhibiting cell size reduction and subsequent enucleation. One of them, alncRNA-EC7, is transcribed from an enhancer and is specifically needed for activation of the neighboring gene encoding BAND 3. Our study provides an annotated catalog of erythroid lncRNAs, readily available through an online resource, and shows that diverse types of lncRNAs participate in the regulatory circuitry underlying erythropoiesis.
Figures
Comment in
-
Long noncoding RNAs in erythropoiesis.Blood. 2014 Jan 23;123(4):465-6. doi: 10.1182/blood-2013-12-538306. Blood. 2014. PMID: 24458276
References
-
- Cantor AB, Orkin SH. Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene. 2002;21(21):3368–3376. - PubMed
-
- Bertone P, Stolc V, Royce TE, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306(5705):2242–2246. - PubMed
-
- Carninci P, Kasukawa T, Katayama S, et al. FANTOM Consortium; RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group) The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–1563. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
