

Channelopathy pathogenesis in autism spectrum disorders
- PMID: 24204377
- PMCID: PMC3817418
- DOI: 10.3389/fgene.2013.00222
Channelopathy pathogenesis in autism spectrum disorders
Abstract
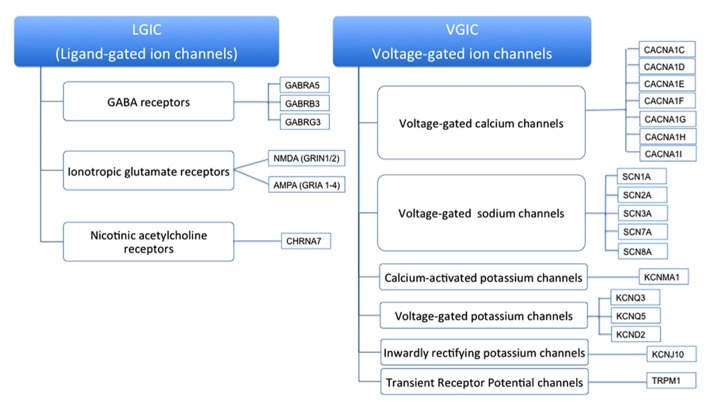
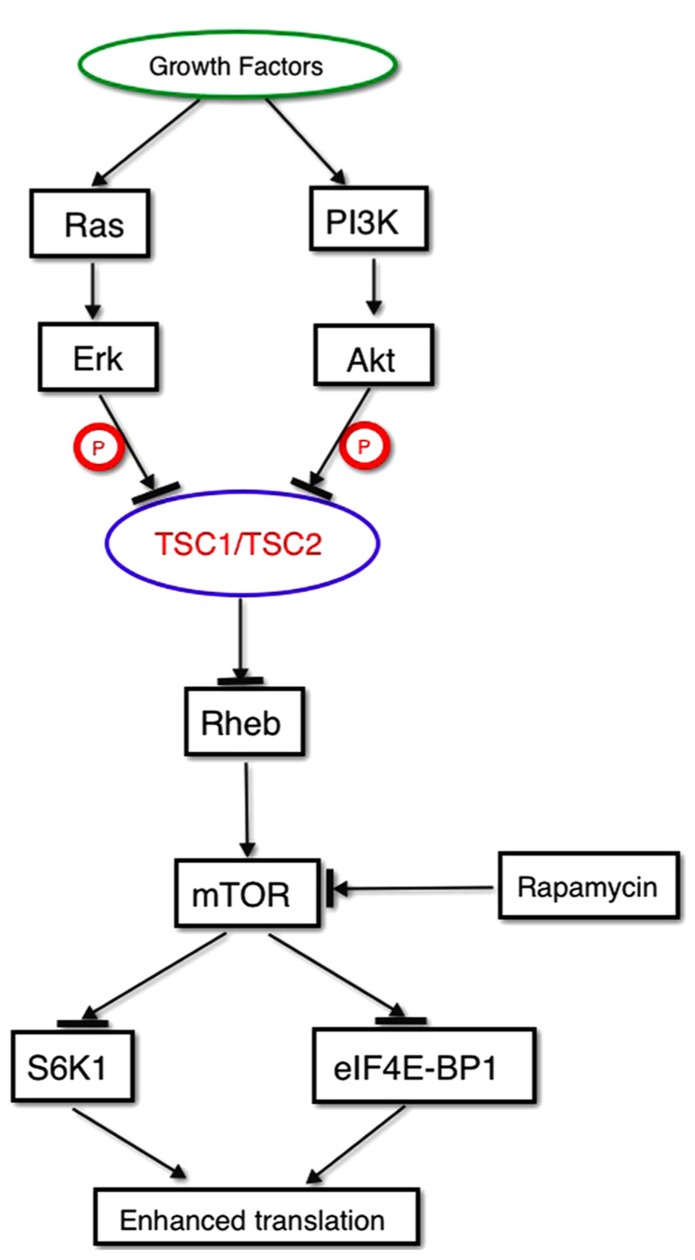
Autism spectrum disorder (ASD) is a syndrome that affects normal brain development and is characterized by impaired social interaction as well as verbal and non-verbal communication and by repetitive, stereotypic behavior. ASD is a complex disorder arising from a combination of multiple genetic and environmental factors that are independent from racial, ethnic and socioeconomical status. The high heritability of ASD suggests a strong genetic basis for the disorder. Furthermore, a mounting body of evidence implies a role of various ion channel gene defects (channelopathies) in the pathogenesis of autism. Indeed, recent genome-wide association, and whole exome- and whole-genome resequencing studies linked polymorphisms and rare variants in calcium, sodium and potassium channels and their subunits with susceptibility to ASD, much as they do with bipolar disorder, schizophrenia and other neuropsychiatric disorders. Moreover, animal models with these genetic variations recapitulate endophenotypes considered to be correlates of autistic behavior seen in patients. An ion flux across the membrane regulates a variety of cell functions, from generation of action potentials to gene expression and cell morphology, thus it is not surprising that channelopathies have profound effects on brain functions. In the present work, we summarize existing evidence for the role of ion channel gene defects in the pathogenesis of autism with a focus on calcium signaling and its downstream effects.
Keywords: Angelman syndrome; Fragile X syndrome; Prader-Willi syndrome; Rett syndrome; calcium; mTOR; tuberous sclerosis.
Figures
References
-
- Adams C. E., Yonchek J. C., Schulz K. M., Graw S. L., Stitzel J., Teschke P. U., et al. (2012). Reduced Chrna7 expression in mice is associated with decreases in hippocampal markers of inhibitory function: implications for neuropsychiatric diseases. Neuroscience 207 274–28210.1016/j.neuroscience.2012.01.033 - DOI - PMC - PubMed
-
- Antzelevitch C., Pollevick G. D., Cordeiro J. M., Casis O., Sanguinetti M. C., Aizawa Y., et al. (2007). Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115 442–44910.1161/CIRCULATIONAHA.106.668392 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous