

The intertwined roles of transcription and repair proteins
- PMID: 24207023
- PMCID: PMC3919531
- DOI: 10.1016/j.molcel.2013.10.018
The intertwined roles of transcription and repair proteins
Abstract
Transcription is apparently risky business. Its intrinsic mutagenic potential must be kept in check by networks of DNA repair factors that monitor the transcription process to repair DNA lesions that could otherwise compromise transcriptional fidelity and genome integrity. Intriguingly, recent studies point to an even more direct function of DNA repair complexes as coactivators of transcription and the unexpected role of "scheduled" DNA damage/repair at gene promoters. Paradoxically, spontaneous DNA double-strand breaks also induce ectopic transcription that is essential for repair. Thus, transcription, DNA damage, and repair may be more physically and functionally intertwined than previously appreciated.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
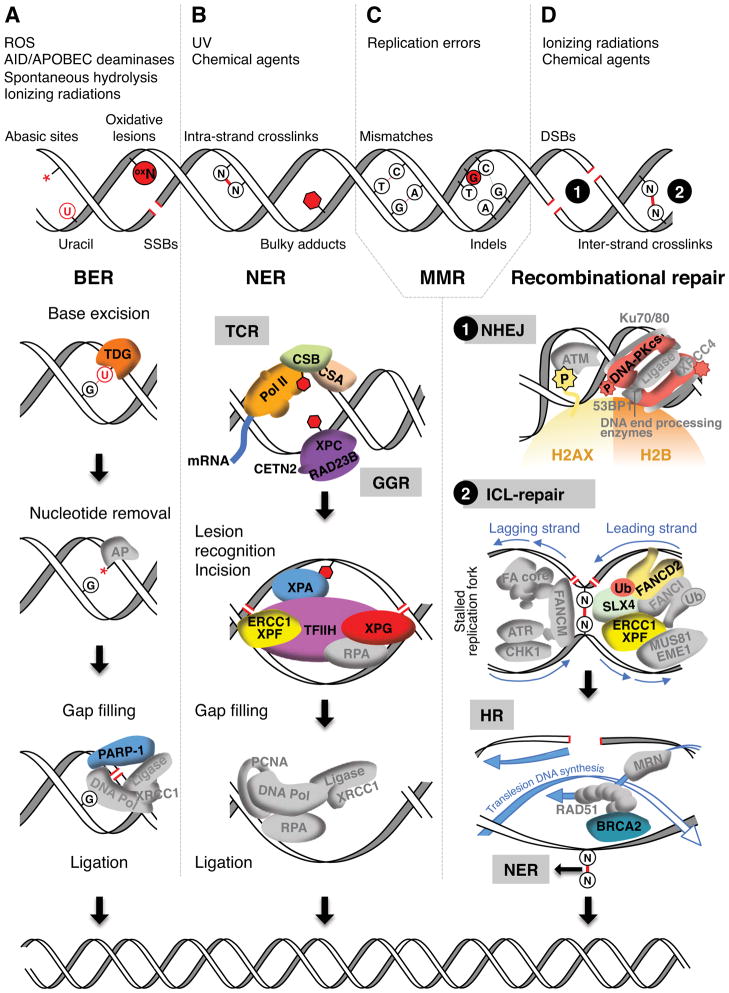
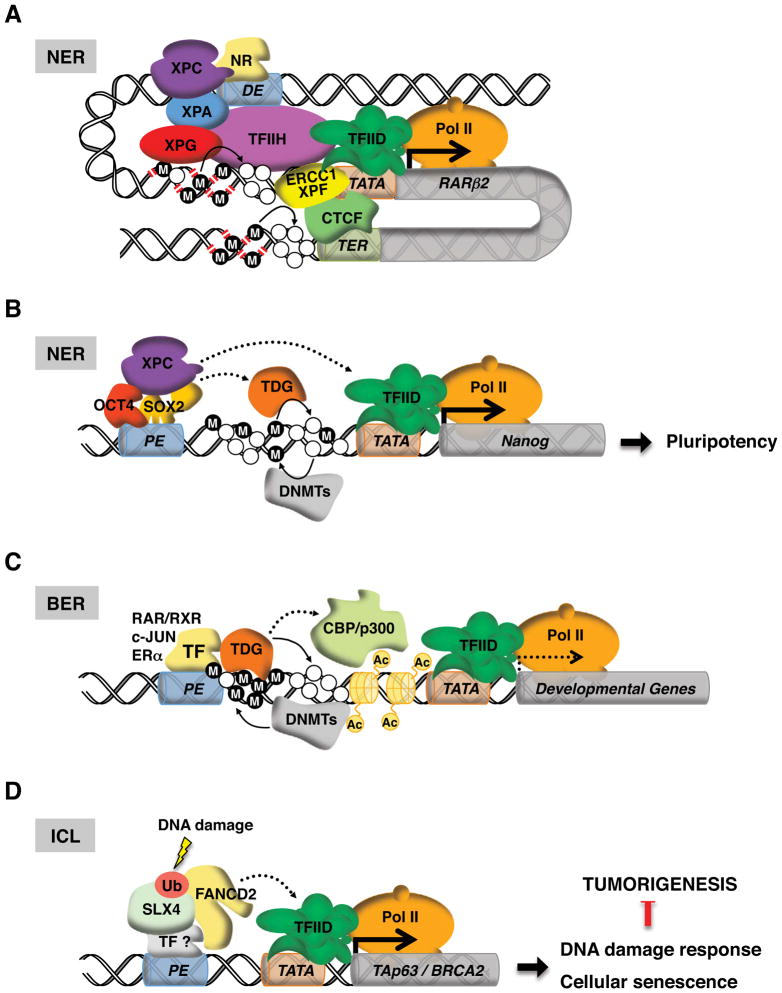
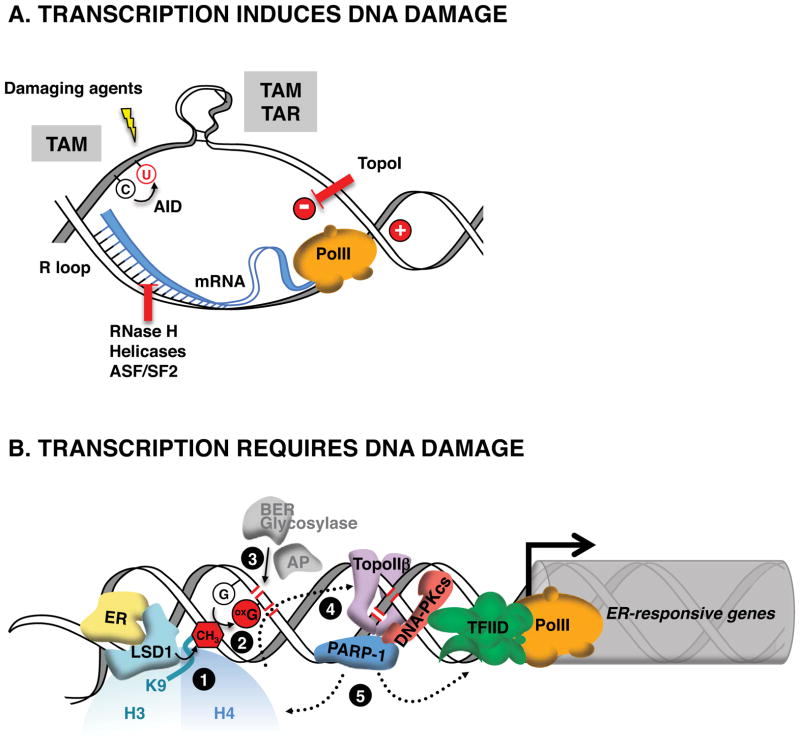
 . The demethylation reaction releases reactive oxygen species that converts nearby guanines (G) into 8-oxo-guanines (oxG)
. The demethylation reaction releases reactive oxygen species that converts nearby guanines (G) into 8-oxo-guanines (oxG)
 . oxG removal by base excision repair (BER) creates DNA nicks
. oxG removal by base excision repair (BER) creates DNA nicks
 that facilitate entrance of the endonuclease topoisomerase IIβ (TopoIIβ)
that facilitate entrance of the endonuclease topoisomerase IIβ (TopoIIβ)
 . TopoIIβ-induced double strand breaks recruit PARP-1 and DNA-PKcs repair enzymes, which induce a permissive chromatin architecture for transcription initiation
. TopoIIβ-induced double strand breaks recruit PARP-1 and DNA-PKcs repair enzymes, which induce a permissive chromatin architecture for transcription initiation
 .
.References
-
- Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Molecular cell. 2012;46:115–124. - PubMed
-
- Araki M, Masutani C, Takemura M, Uchida A, Sugasawa K, Kondoh J, Ohkuma Y, Hanaoka F. Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. The Journal of biological chemistry. 2001;276:18665–18672. - PubMed
-
- Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
