

The landscape of microsatellite instability in colorectal and endometrial cancer genomes
- PMID: 24209623
- PMCID: PMC3871995
- DOI: 10.1016/j.cell.2013.10.015
The landscape of microsatellite instability in colorectal and endometrial cancer genomes
Abstract
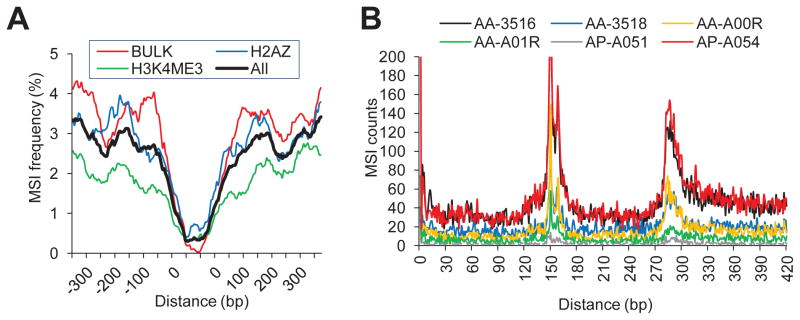
Microsatellites-simple tandem repeats present at millions of sites in the human genome-can shorten or lengthen due to a defect in DNA mismatch repair. We present here a comprehensive genome-wide analysis of the prevalence, mutational spectrum, and functional consequences of microsatellite instability (MSI) in cancer genomes. We analyzed MSI in 277 colorectal and endometrial cancer genomes (including 57 microsatellite-unstable ones) using exome and whole-genome sequencing data. Recurrent MSI events in coding sequences showed tumor type specificity, elevated frameshift-to-inframe ratios, and lower transcript levels than wild-type alleles. Moreover, genome-wide analysis revealed differences in the distribution of MSI versus point mutations, including overrepresentation of MSI in euchromatic and intronic regions compared to heterochromatic and intergenic regions, respectively, and depletion of MSI at nucleosome-occupied sequences. Our results provide a panoramic view of MSI in cancer genomes, highlighting their tumor type specificity, impact on gene expression, and the role of chromatin organization.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures






References
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
