

Differential methylation relative to breast cancer subtype and matched normal tissue reveals distinct patterns
- PMID: 24212716
- PMCID: PMC3832780
- DOI: 10.1007/s10549-013-2738-0
Differential methylation relative to breast cancer subtype and matched normal tissue reveals distinct patterns
Abstract
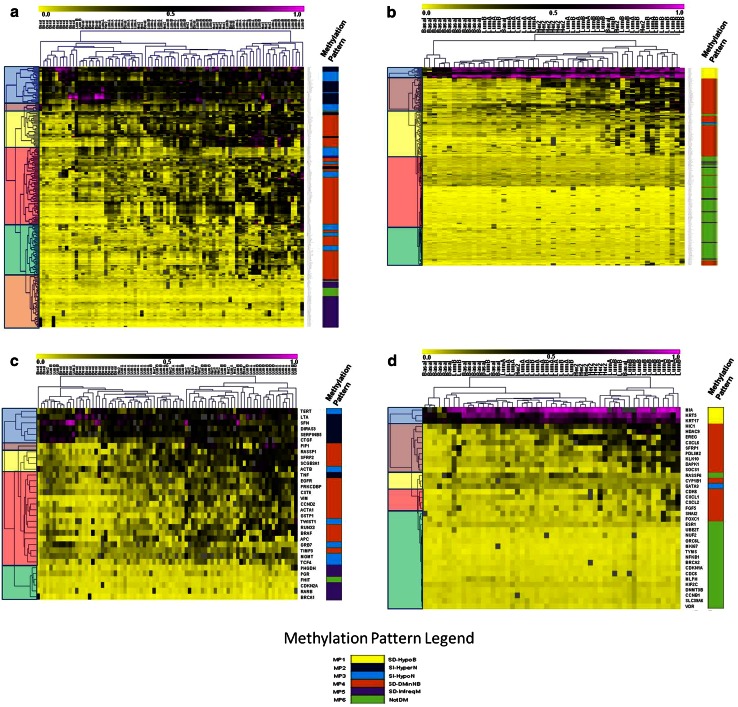
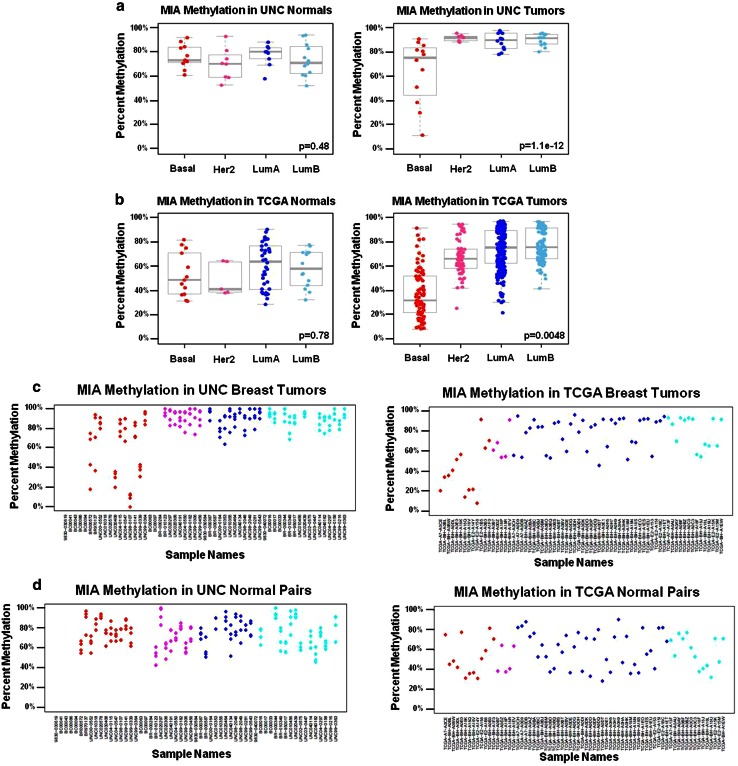
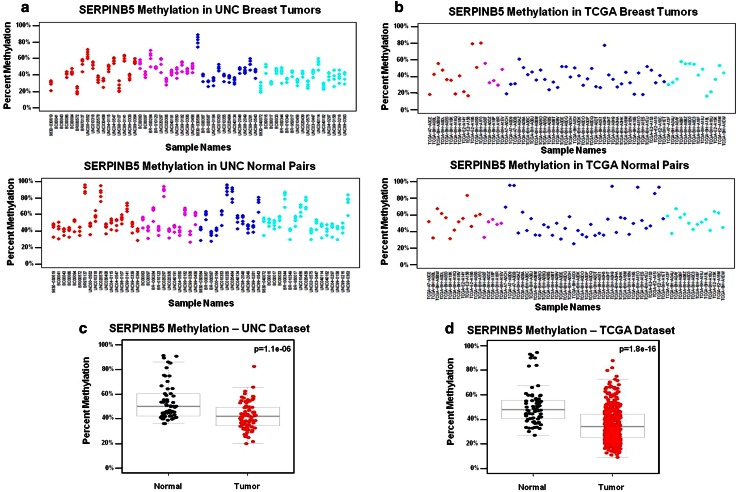
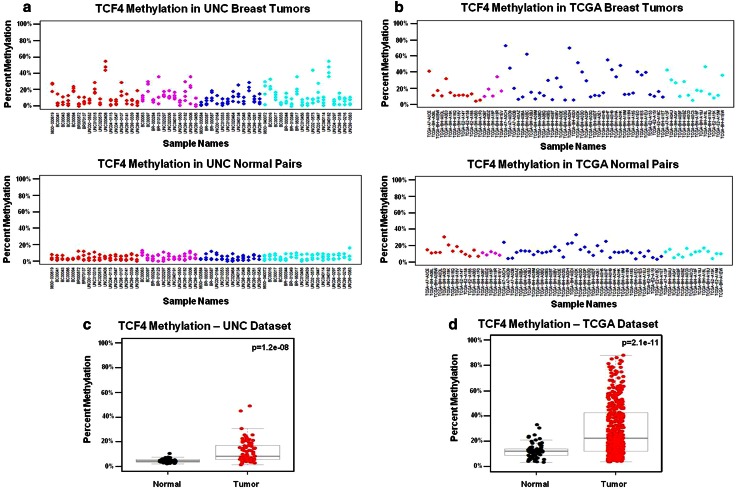
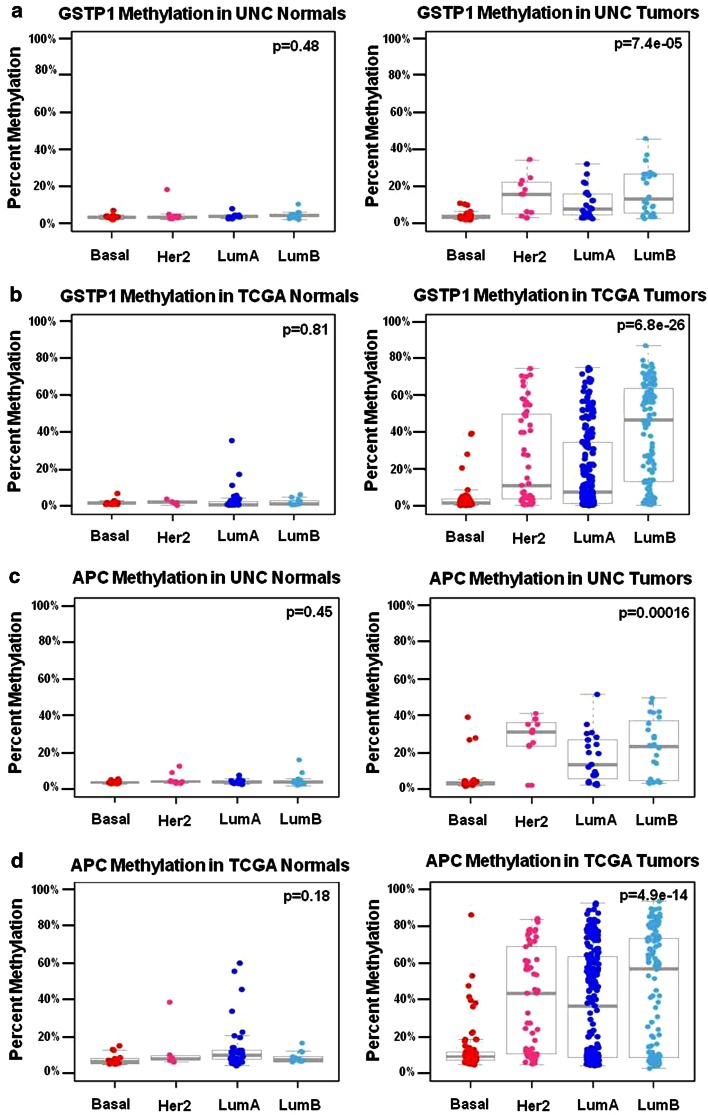
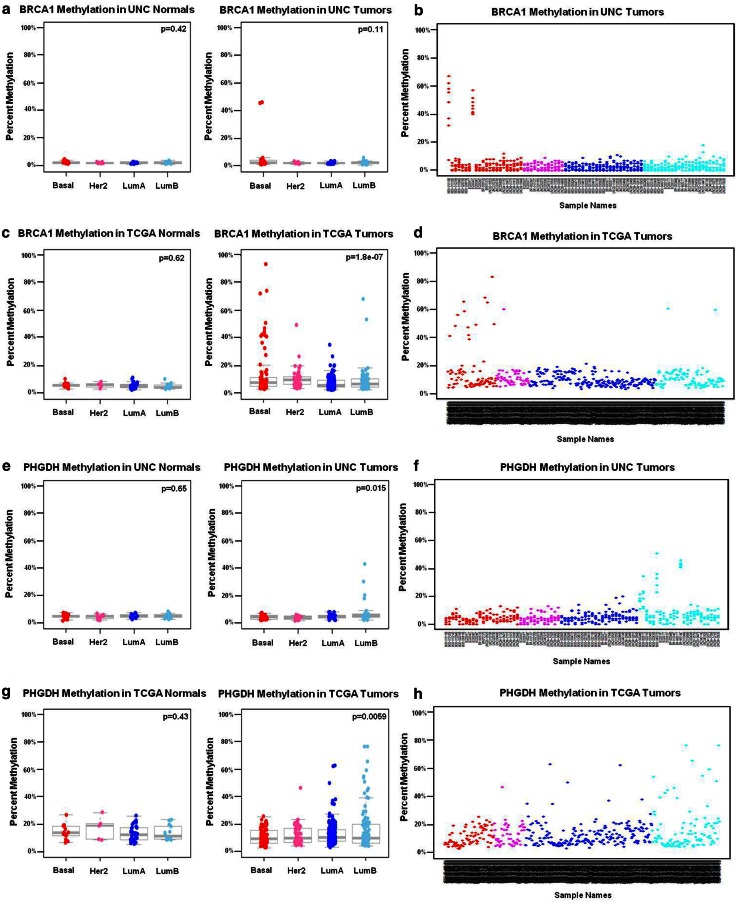
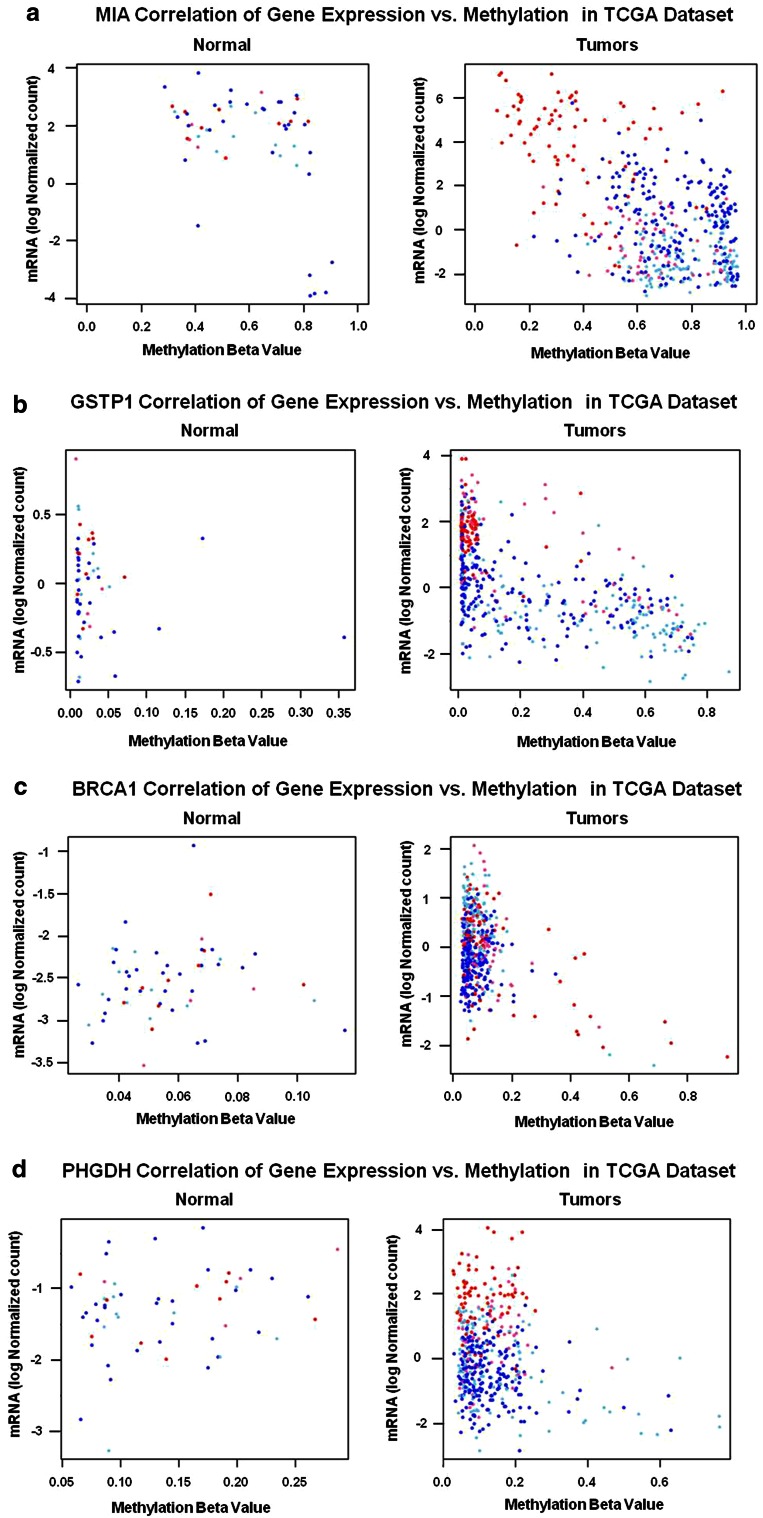
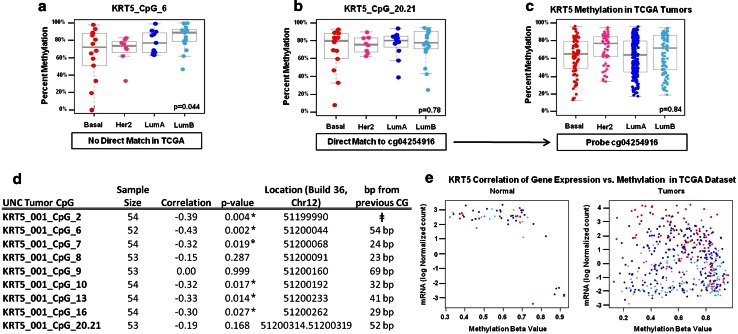
Due to the heterogeneous nature of breast cancer and the widespread use of single-gene studies, there is limited knowledge of multi-gene, locus-specific DNA methylation patterns in relation to molecular subtype and clinical features. We, therefore, quantified DNA methylation of 70 candidate gene loci in 140 breast tumors and matched normal tissues and determined associations with gene expression and tumor subtype. Using Sequenom's EpiTYPER platform, approximately 1,200 CpGs were interrogated and revealed six DNA methylation patterns in breast tumors relative to matched normal tissue. Differential methylation of several gene loci was observed within all molecular subtypes, while other patterns were subtype-dependent. Methylation of numerous gene loci was inversely correlated with gene expression, and in some cases, this correlation was only observed within specific breast tumor subtypes. Our findings were validated on a larger set of tumors and matched adjacent normal tissue from The Cancer Genome Atlas dataset, which utilized methylation data derived from both Illumina Infinium 27 and 450 k arrays. These findings highlight the need to control for subtype when interpreting DNA methylation results, and the importance of interrogating multiple CpGs across varied gene regions.
Figures
References
-
- Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nature reviews. Cancer. 2004;4:707–717. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
