

HCV infection selectively impairs type I but not type III IFN signaling
- PMID: 24215913
- PMCID: PMC3873489
- DOI: 10.1016/j.ajpath.2013.10.005
HCV infection selectively impairs type I but not type III IFN signaling
Abstract
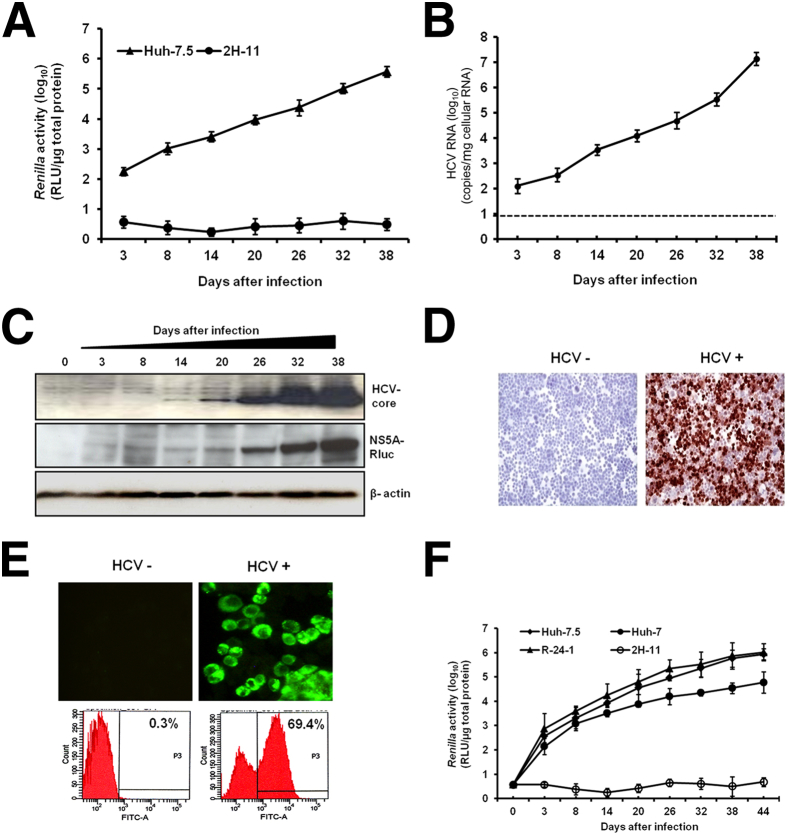
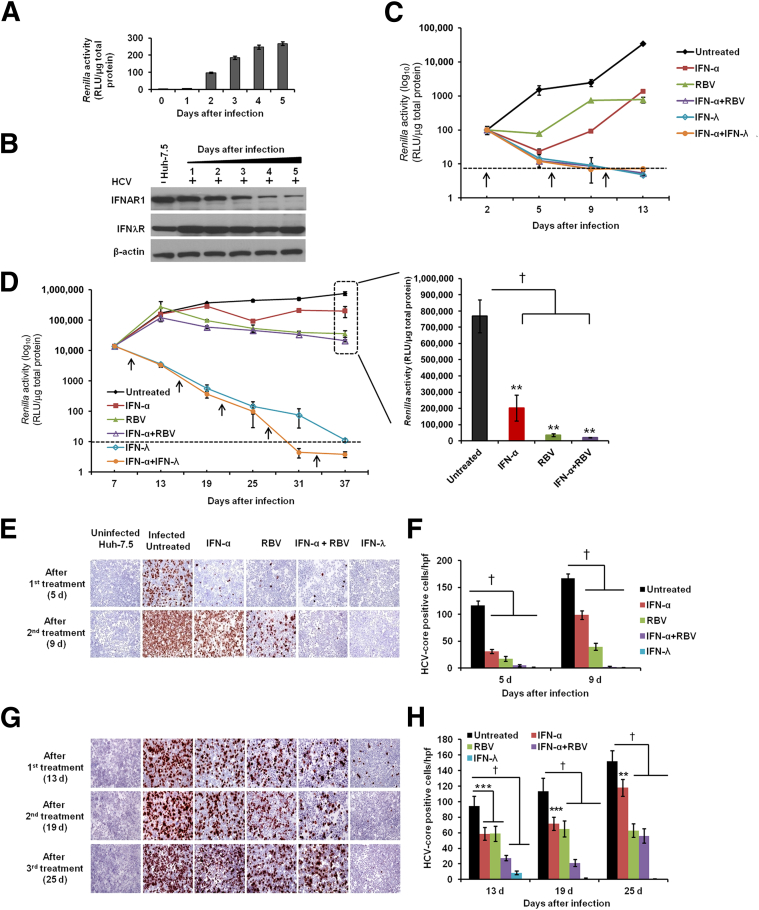
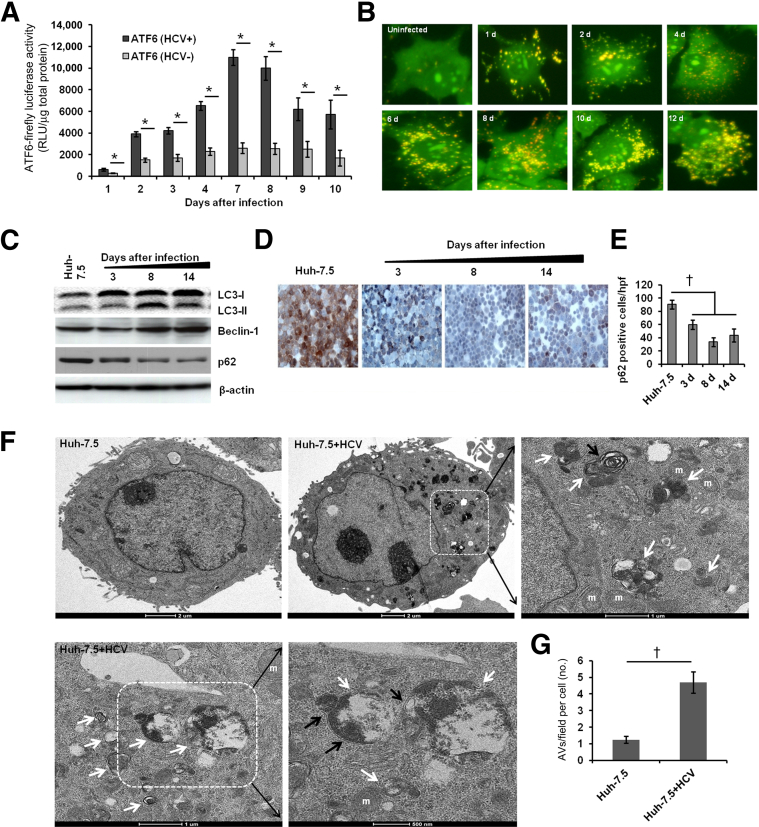
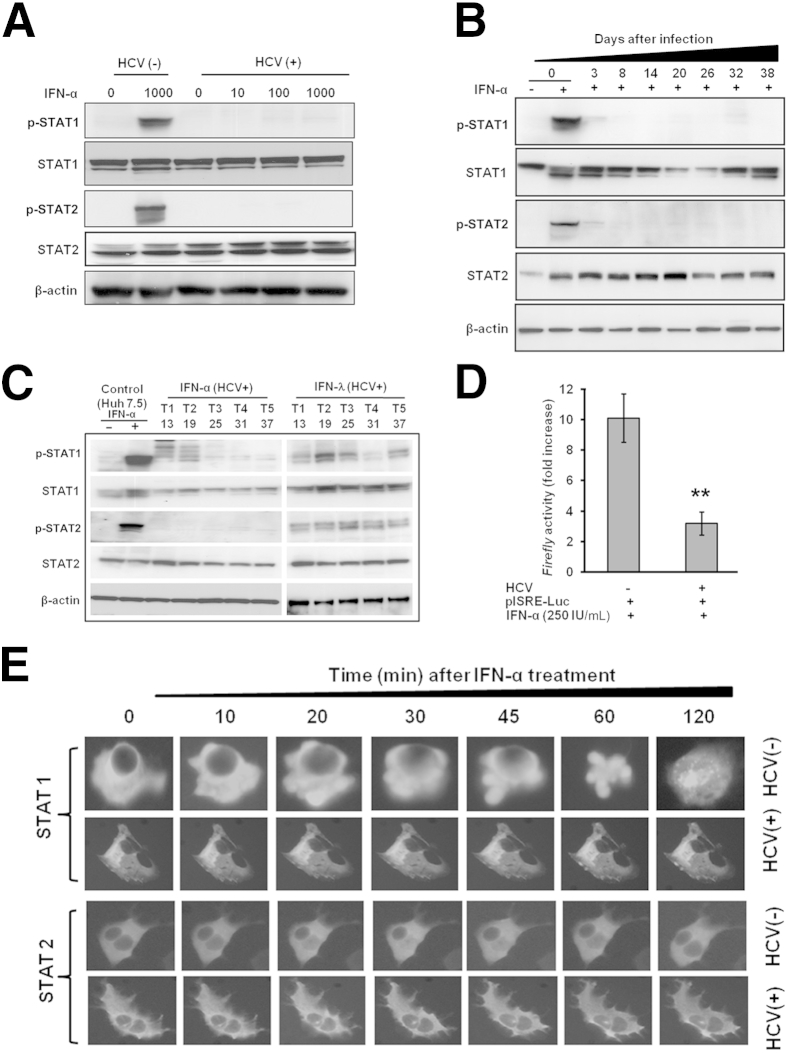
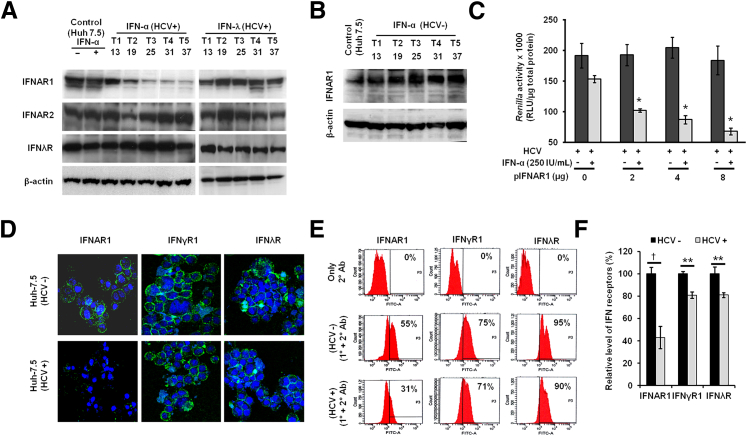
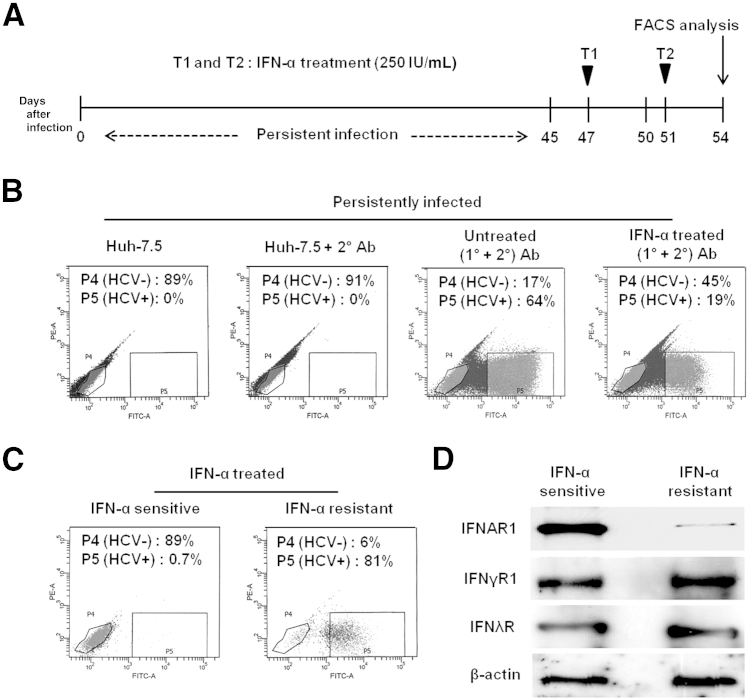
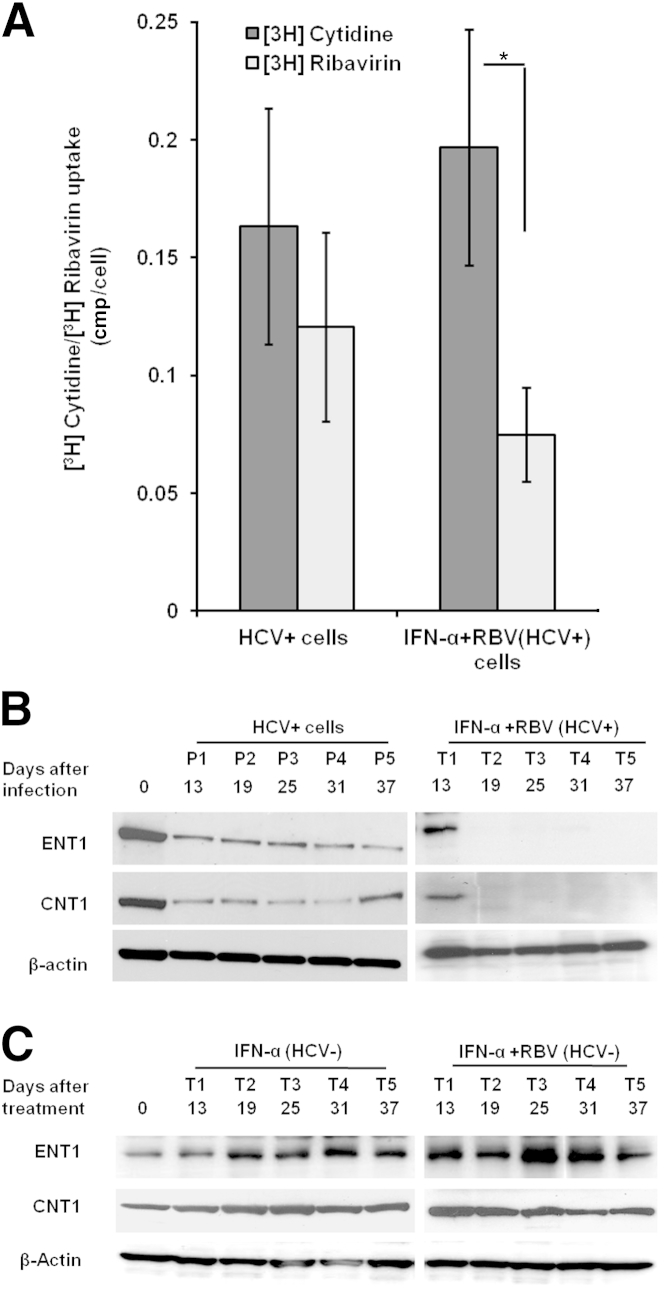
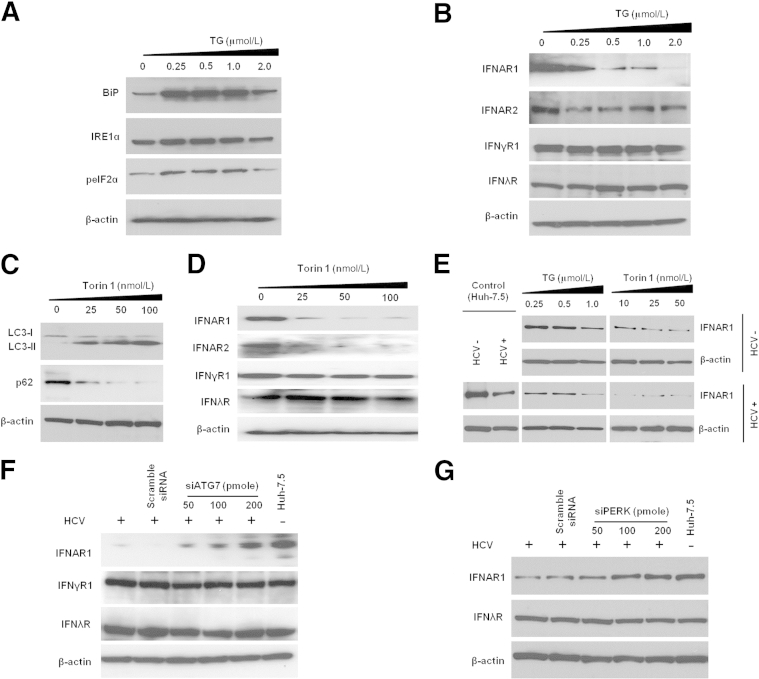
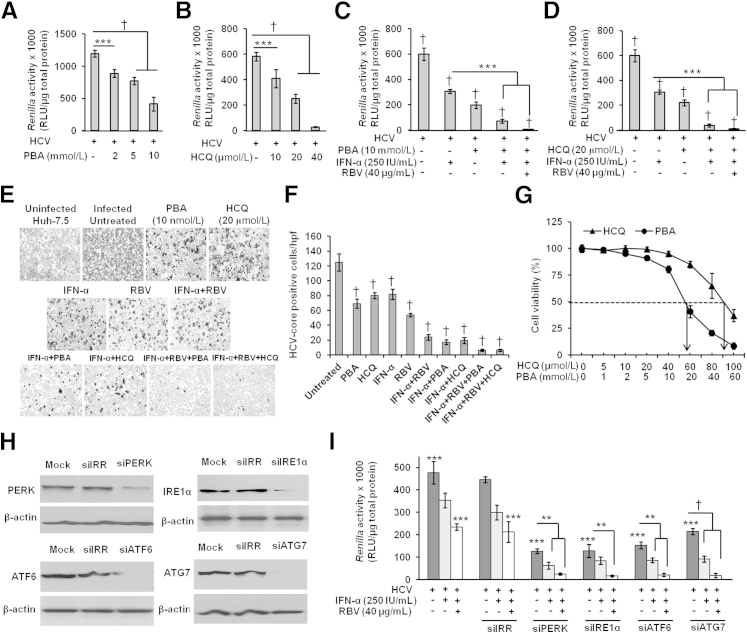
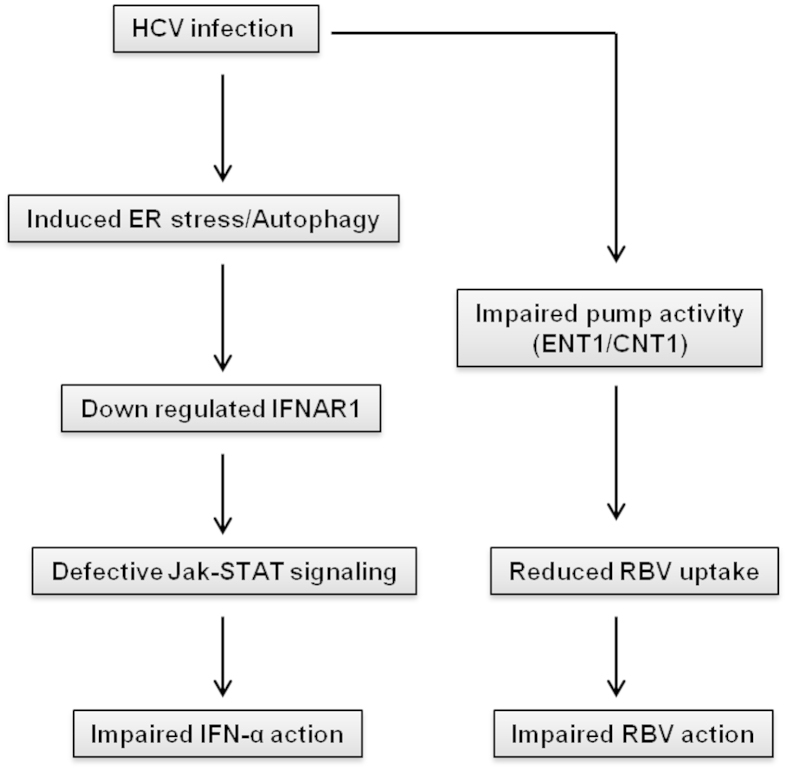
A stable and persistent Hepatitis C virus (HCV) replication cell culture model was developed to examine clearance of viral replication during long-term treatment using interferon-α (IFN-α), IFN-λ, and ribavirin (RBV). Persistently HCV-infected cell culture exhibited an impaired antiviral response to IFN-α+RBV combination treatment, whereas IFN-λ treatment produced a strong and sustained antiviral response that cleared HCV replication. HCV replication in persistently infected cells induced chronic endoplasmic reticulum (ER) stress and an autophagy response that selectively down-regulated the functional IFN-α receptor-1 chain of type I, but not type II (IFN-γ) or type III (IFN-λ) IFN receptors. Down-regulation of IFN-α receptor-1 resulted in defective JAK-STAT signaling, impaired STAT phosphorylation, and impaired nuclear translocation of STAT. Furthermore, HCV replication impaired RBV uptake, because of reduced expression of the nucleoside transporters ENT1 and CNT1. Silencing ER stress and the autophagy response using chemical inhibitors or siRNA additively inhibited HCV replication and induced viral clearance by the IFN-α+RBV combination treatment. These results indicate that HCV induces ER stress and that the autophagy response selectively impairs type I (but not type III) IFN signaling, which explains why IFN-λ (but not IFN-α) produced a sustained antiviral response against HCV. The results also indicate that inhibition of ER stress and of the autophagy response overcomes IFN-α+RBV resistance mechanisms associated with HCV infection.
Copyright © 2014 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Shepard C.W., Finelli L., Alter M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–567. - PubMed
-
- Butt A.A., Kanwal F. Boceprevir and telaprevir in the management of hepatitis C virus-infected patients. Clin Infect Dis. 2012;54:96–104. - PubMed
-
- Dore G.J., Matthews G.V., Rockstroh J. Future of hepatitis C therapy: development of direct-acting antivirals. Curr Opin HIV AIDS. 2011;6:508–513. - PubMed
-
- Halfon P., Locarnini S.J. Hepatitis C virus resistance to protease inhibitors. Hepatology. 2011;55:192–206. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
