

Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo
- PMID: 24218140
- PMCID: PMC3832937
- DOI: 10.1084/jem.20131141
Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo
Abstract
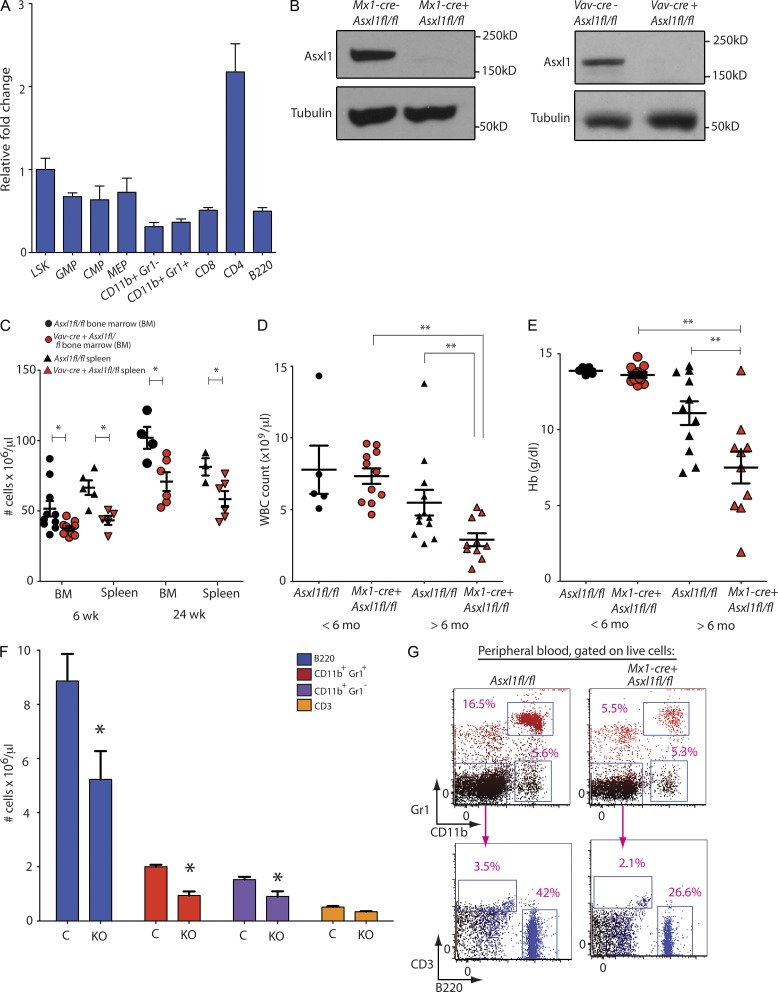
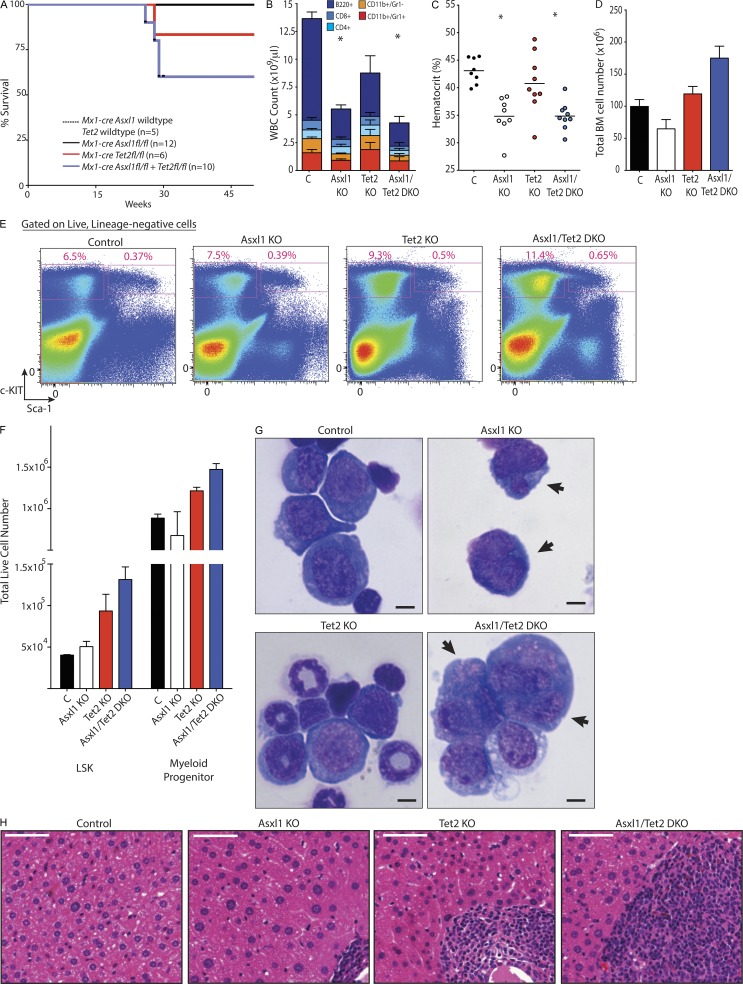
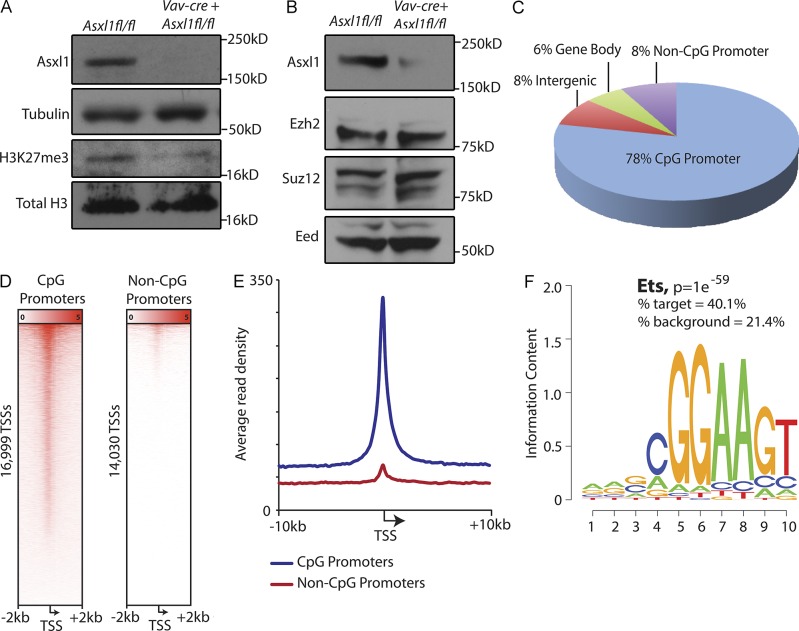
Somatic Addition of Sex Combs Like 1 (ASXL1) mutations occur in 10-30% of patients with myeloid malignancies, most commonly in myelodysplastic syndromes (MDSs), and are associated with adverse outcome. Germline ASXL1 mutations occur in patients with Bohring-Opitz syndrome. Here, we show that constitutive loss of Asxl1 results in developmental abnormalities, including anophthalmia, microcephaly, cleft palates, and mandibular malformations. In contrast, hematopoietic-specific deletion of Asxl1 results in progressive, multilineage cytopenias and dysplasia in the context of increased numbers of hematopoietic stem/progenitor cells, characteristic features of human MDS. Serial transplantation of Asxl1-null hematopoietic cells results in a lethal myeloid disorder at a shorter latency than primary Asxl1 knockout (KO) mice. Asxl1 deletion reduces hematopoietic stem cell self-renewal, which is restored by concomitant deletion of Tet2, a gene commonly co-mutated with ASXL1 in MDS patients. Moreover, compound Asxl1/Tet2 deletion results in an MDS phenotype with hastened death compared with single-gene KO mice. Asxl1 loss results in a global reduction of H3K27 trimethylation and dysregulated expression of known regulators of hematopoiesis. RNA-Seq/ChIP-Seq analyses of Asxl1 in hematopoietic cells identify a subset of differentially expressed genes as direct targets of Asxl1. These findings underscore the importance of Asxl1 in Polycomb group function, development, and hematopoiesis.
Figures






References
-
- Bejar R., Stevenson K.E., Caughey B.A., Abdel-Wahab O., Steensma D.P., Galili N., Raza A., Kantarjian H., Levine R.L., Neuberg D., et al. 2012. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 30:3376–3382 10.1200/JCO.2011.40.7379 - DOI - PMC - PubMed
-
- Boultwood J., Perry J., Pellagatti A., Fernandez-Mercado M., Fernandez-Santamaria C., Calasanz M.J., Larrayoz M.J., Garcia-Delgado M., Giagounidis A., Malcovati L., et al. 2010. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 24:1062–1065 10.1038/leu.2010.20 - DOI - PubMed
-
- Bracken A.P., Kleine-Kohlbrecher D., Dietrich N., Pasini D., Gargiulo G., Beekman C., Theilgaard-Mönch K., Minucci S., Porse B.T., Marine J.C., et al. 2007. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21:525–530 10.1101/gad.415507 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 1R01CA173636/CA/NCI NIH HHS/United States
- P30 CA016087/CA/NCI NIH HHS/United States
- R01 CA133379/CA/NCI NIH HHS/United States
- U01 HL100395/HL/NHLBI NIH HHS/United States
- R01CA133379/CA/NCI NIH HHS/United States
- K08 CA160647/CA/NCI NIH HHS/United States
- R01 CA149655/CA/NCI NIH HHS/United States
- 5R01CA173636/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01CA149655/CA/NCI NIH HHS/United States
- R01 CA138234/CA/NCI NIH HHS/United States
- R01 CA173636/CA/NCI NIH HHS/United States
- T32 CA009161/CA/NCI NIH HHS/United States
- 5U01HL100395/HL/NHLBI NIH HHS/United States
- R01 CA105129/CA/NCI NIH HHS/United States
- 5R01CA173636-01/CA/NCI NIH HHS/United States
- 1R01CA138234-01/CA/NCI NIH HHS/United States
- R01 CA169784/CA/NCI NIH HHS/United States
- 1K08CA160647-01/CA/NCI NIH HHS/United States
- R01CA105129/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
