

From prion diseases to prion-like propagation mechanisms of neurodegenerative diseases
- PMID: 24222767
- PMCID: PMC3810426
- DOI: 10.1155/2013/975832
From prion diseases to prion-like propagation mechanisms of neurodegenerative diseases
Abstract
Prion diseases are fatal neurodegenerative sporadic, inherited, or acquired disorders. In humans, Creutzfeldt-Jakob disease is the most studied prion disease. In animals, the most frequent prion diseases are scrapie in sheep and goat, bovine spongiform encephalopathy in cattle, and the emerging chronic wasting disease in wild and captive deer in North America. The hallmark of prion diseases is the deposition in the brain of PrP(Sc), an abnormal β -sheet-rich form of the cellular prion protein (PrP(C)) (Prusiner 1982). According to the prion hypothesis, PrP(Sc) can trigger the autocatalytic conversion of PrP(C) into PrP(Sc), presumably in the presence of cofactors (lipids and small RNAs) that have been recently identified. In this review, we will come back to the original works that led to the discovery of prions and to the protein-only hypothesis proposed by Dr. Prusiner. We will then describe the recent reports on mammalian synthetic prions and recombinant prions that strongly support the protein-only hypothesis. The new concept of "deformed templating" regarding a new mechanism of PrP(Sc) formation and replication will be exposed. The review will end with a chapter on the prion-like propagation of other neurodegenerative disorders, such as Alzheimer's and Parkinson's disease and tauopathies.
Figures
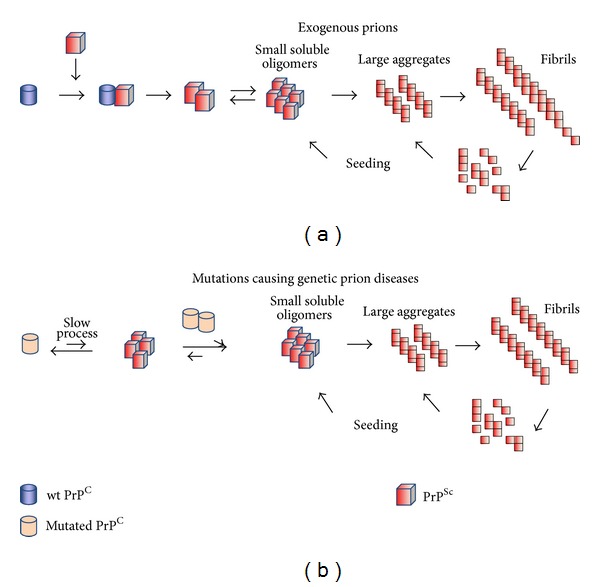
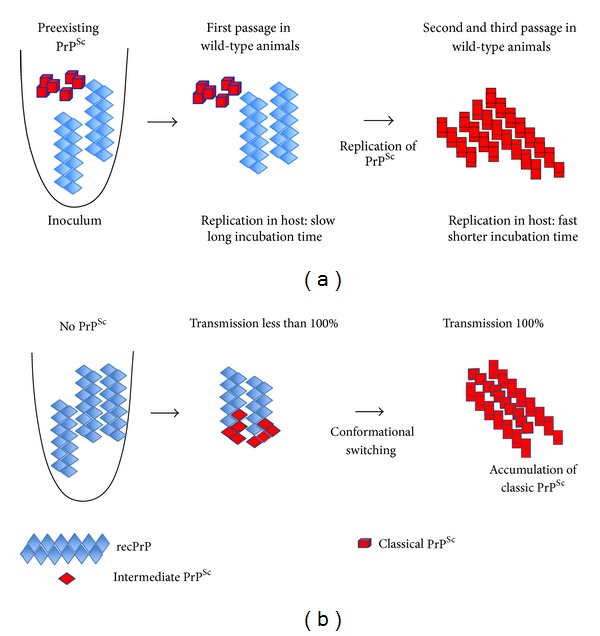
References
-
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. - PubMed
-
- Dormont D. Prion diseases: pathogenesis and public health concerns. FEBS Letters. 2002;529(1):17–21. - PubMed
-
- Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. The Lancet. 1996;347(9006):921–925. - PubMed
-
- Peden AH, Head MW, Ritchie DL, Bell PJE, Ironside PJW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. The Lancet. 2004;364(9433):527–529. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
