

Evolution and diversity in human herpes simplex virus genomes
- PMID: 24227835
- PMCID: PMC3911644
- DOI: 10.1128/JVI.01987-13
Evolution and diversity in human herpes simplex virus genomes
Abstract
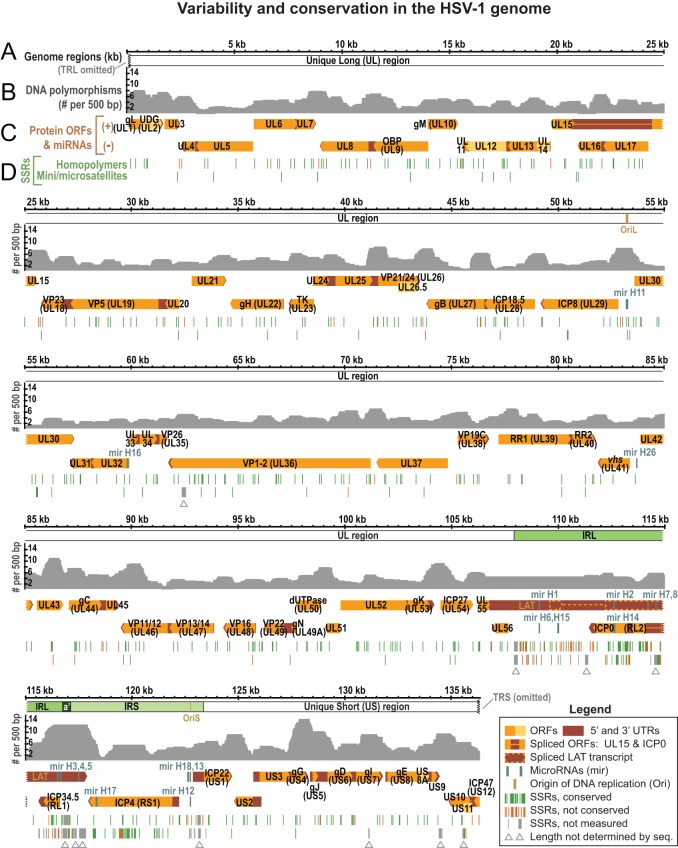
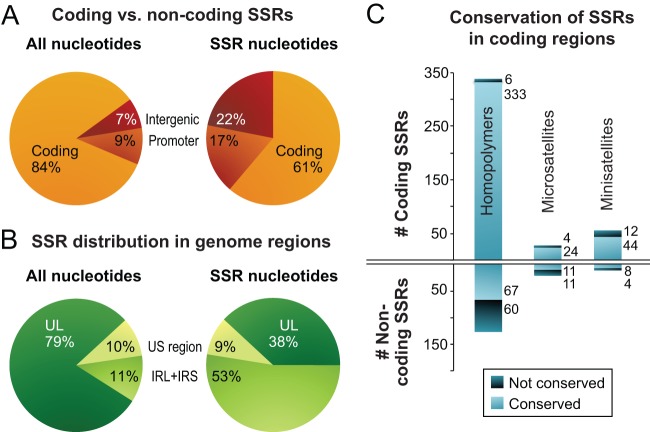
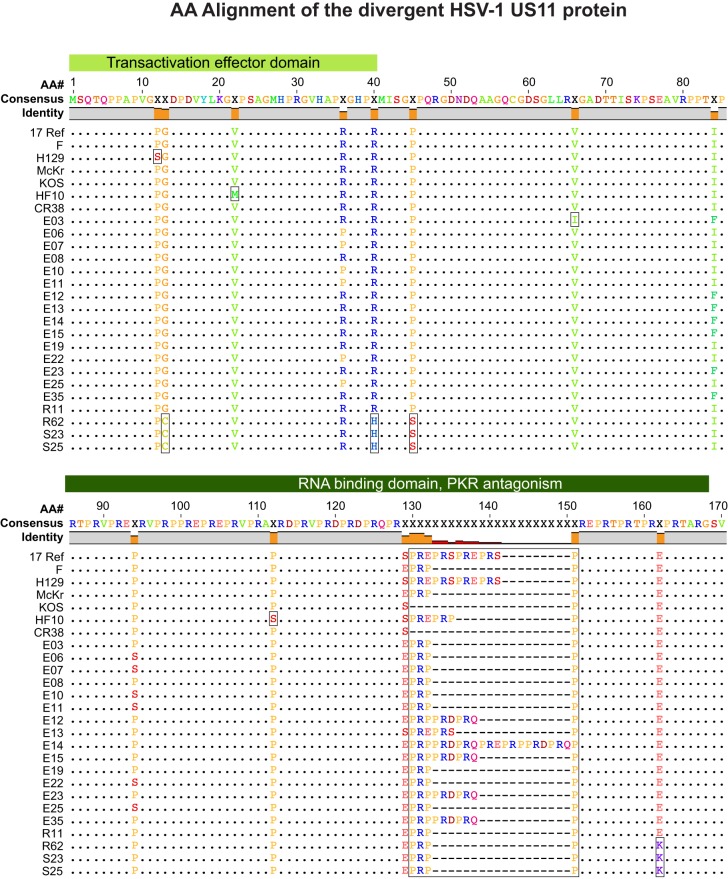
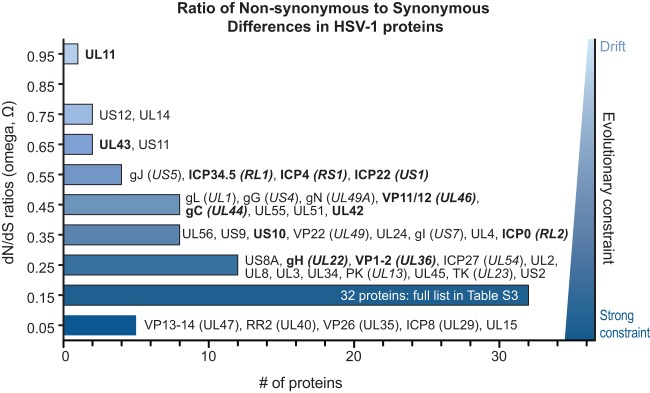
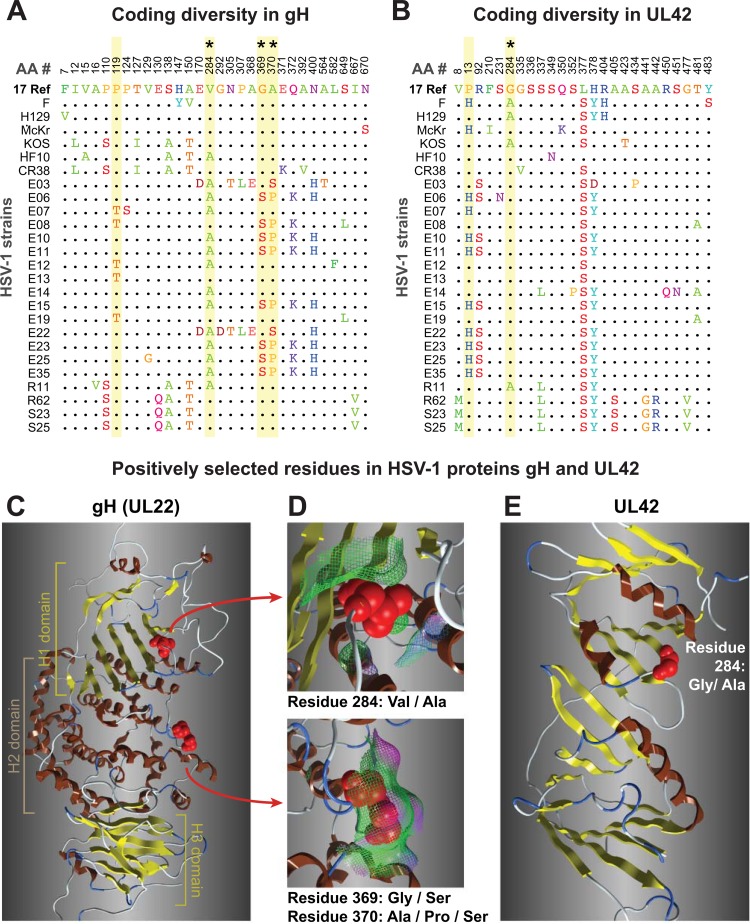
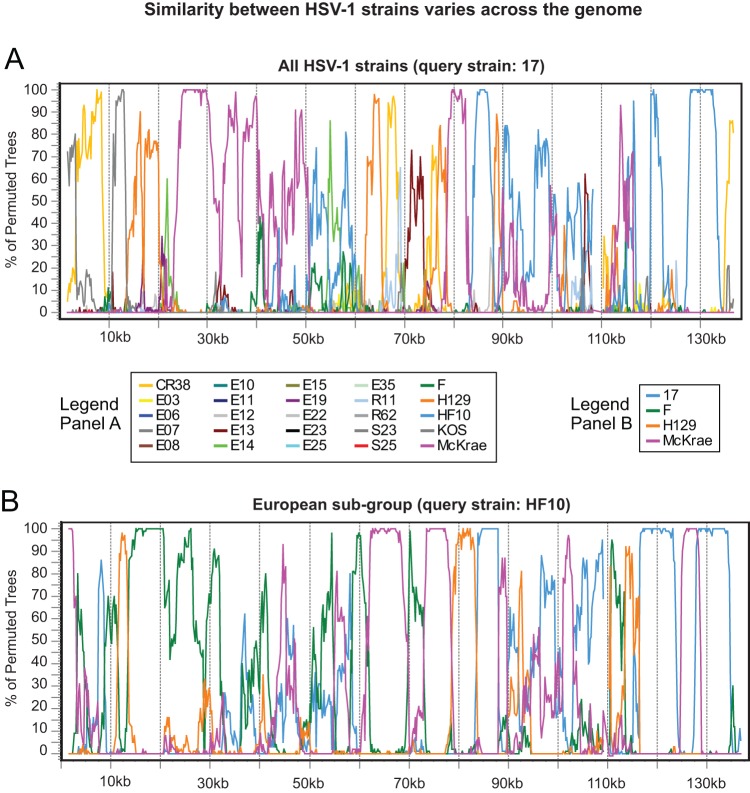
Herpes simplex virus 1 (HSV-1) causes a chronic, lifelong infection in >60% of adults. Multiple recent vaccine trials have failed, with viral diversity likely contributing to these failures. To understand HSV-1 diversity better, we comprehensively compared 20 newly sequenced viral genomes from China, Japan, Kenya, and South Korea with six previously sequenced genomes from the United States, Europe, and Japan. In this diverse collection of passaged strains, we found that one-fifth of the newly sequenced members share a gene deletion and one-third exhibit homopolymeric frameshift mutations (HFMs). Individual strains exhibit genotypic and potential phenotypic variation via HFMs, deletions, short sequence repeats, and single-nucleotide polymorphisms, although the protein sequence identity between strains exceeds 90% on average. In the first genome-scale analysis of positive selection in HSV-1, we found signs of selection in specific proteins and residues, including the fusion protein glycoprotein H. We also confirmed previous results suggesting that recombination has occurred with high frequency throughout the HSV-1 genome. Despite this, the HSV-1 strains analyzed clustered by geographic origin during whole-genome distance analysis. These data shed light on likely routes of HSV-1 adaptation to changing environments and will aid in the selection of vaccine antigens that are invariant worldwide.
Figures



Comment in
-
Korean war and the origin of herpes simplex virus 1 strain KOS.J Virol. 2014 Apr;88(7):3911. doi: 10.1128/JVI.00010-14. Epub 2014 Jan 15. J Virol. 2014. PMID: 24429358 Free PMC article. No abstract available.
References
-
- Roizman B, Sears E. 1996. Herpes simplex viruses and their replication, p 1043–1107 InIn Fields BN, Knipe DM, Howley PM. (ed), Fundamental virology, 3rd ed. Lippincott-Raven, Philadelphia, PA
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
