

Evidence for prion-like mechanisms in several neurodegenerative diseases: potential implications for immunotherapy
- PMID: 24228054
- PMCID: PMC3817797
- DOI: 10.1155/2013/473706
Evidence for prion-like mechanisms in several neurodegenerative diseases: potential implications for immunotherapy
Abstract
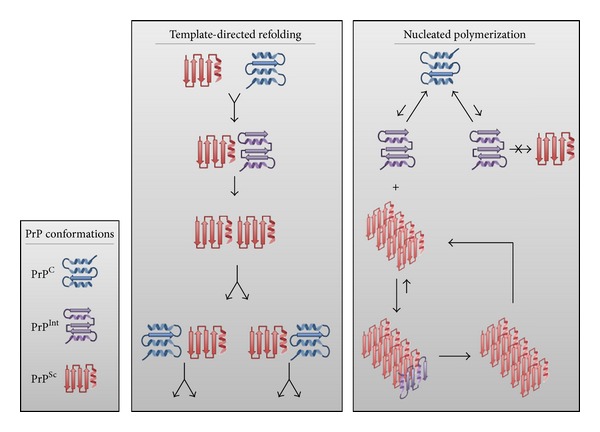
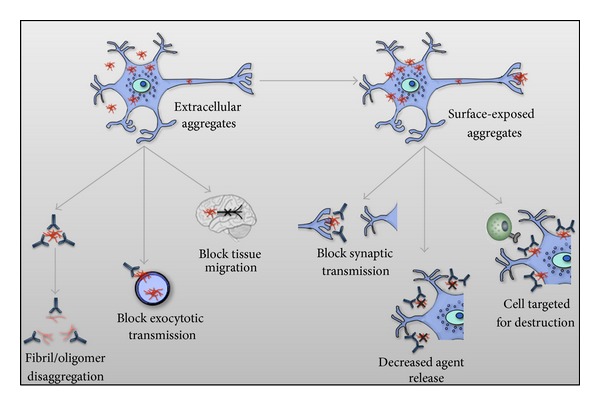
Transmissible spongiform encephalopathies (TSEs) are fatal, untreatable neurodegenerative diseases. While the impact of TSEs on human health is relatively minor, these diseases are having a major influence on how we view, and potentially treat, other more common neurodegenerative disorders. Until recently, TSEs encapsulated a distinct category of neurodegenerative disorder, exclusive in their defining characteristic of infectivity. It now appears that similar mechanisms of self-propagation may underlie other proteinopathies such as Alzheimer's disease, Parkinson's disease, Amyotrophic lateral sclerosis, and Huntington's disease. This link is of scientific interest and potential therapeutic importance as this route of self-propagation offers conceptual support and guidance for vaccine development efforts. Specifically, the existence of a pathological, self-promoting isoform offers a rational vaccine target. Here, we review the evidence of prion-like mechanisms within a number of common neurodegenerative disorders and speculate on potential implications and opportunities for vaccine development.
Figures
References
-
- Silveira JR, Caughey B, Baron GS. Mad Cow Disease and Related Spongiform Encephalopathies. Vol. 284. Berlin, Germany: Springer; 2004. Prion protein and the molecular features of transmissible spongiform encephalopathy agents; pp. 1–50. - PubMed
-
- Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature. 1997;389(6650):498–501. - PubMed
-
- Hill AF, Desbruslais M, Joiner S, et al. The same prion strain causes vCJD and BSE. Nature. 1997;389(6650):448–450. - PubMed
-
- Raymonds GJ, Hope J, Kocisko DA, et al. Molecular assessment of the potential transmissibilities of BSE and scrapie to humans. Nature. 1997;388(6639):285–288. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
