

Review
doi: 10.1016/j.ccr.2013.09.015.
Histone H3.3 mutations: a variant path to cancer
Affiliations
- PMID: 24229707
- PMCID: PMC3882088
- DOI: 10.1016/j.ccr.2013.09.015
Item in Clipboard
Review
Histone H3.3 mutations: a variant path to cancer
Cancer Cell.
.
Abstract
A host of cancer types exhibit aberrant histone modifications. Recently, distinct and recurrent mutations in a specific histone variant, histone H3.3, have been implicated in a high proportion of malignant pediatric brain cancers. The presence of mutant H3.3 histone disrupts epigenetic posttranslational modifications near genes involved in cancer processes and in brain function. Here, we review possible mechanisms by which mutant H3.3 histones may act to promote tumorigenesis. Furthermore, we discuss how perturbations in normal H3.3 chromatin-related and epigenetic functions may more broadly contribute to the formation of human cancers.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
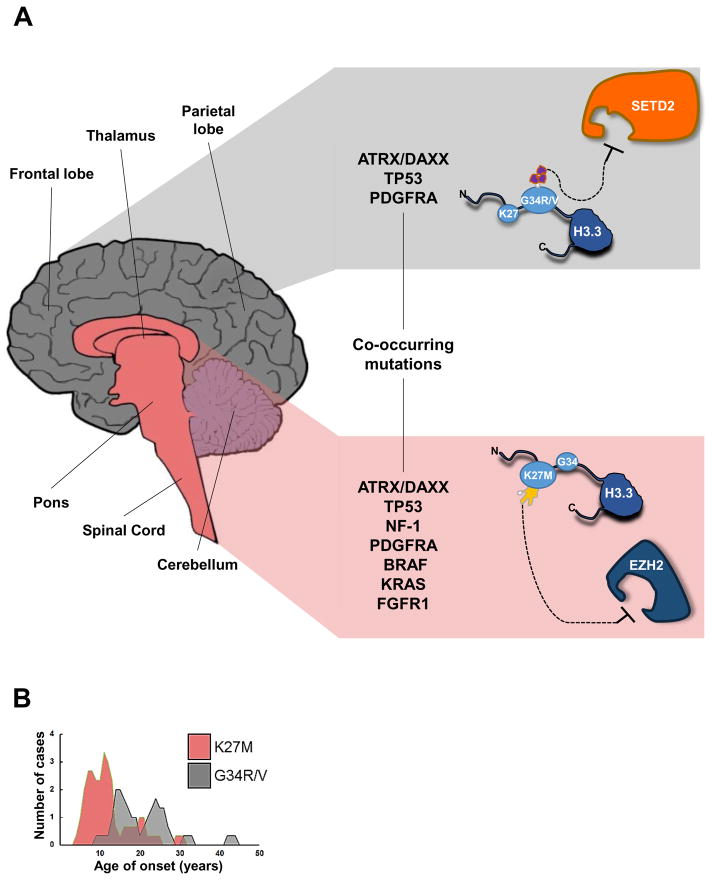
G34R/V and K27M mutations of H3F3A display distinct and independent characteristics from one another. (A) Characteristics of H3.3-mutated tumors. G34R/V mutations (gray, top) in H3F3A localize primarily to cerebral/cortical hemispheres, specifically in frontal, parietal, occipital, and temporal lobes. K27M mutations (pink, bottom) in H3F3A localize primarily to midline locations, including the spinal cord, thalamus, pons, and brainstem. G34R/V mutations significantly overlap with mutations in TP53 and ATRX/DAXX, but are also found simultaneously at low rates with mutations in PDGFRA. K27M mutations overlap with mutations in TP53 and ATRX/DAXX, although not at significantly different rates over control groups. In addition, K27M mutations are found simultaneously at low rates with mutations in NF-1, PDGFRA, BRAF, KRAS, and FGFR1. Mutations in H3.3 directly (K27M) or indirectly (G34R/V) alter post-translationally modified residues. G34R/V mutations appear to affect K36me3 levels, possibly through inhibition of the methyltransferase SETD2, while K27M mutations attenuate EZH2 methyltransferase function, decreasing global K27me3 levels. (B). Age of onset of H3.3-mutated tumors. K27M mutations are more prevalent in younger patients (median age 11 years) while G34R/V mutations are more prevalent in older patients (median age 20 years). The age distribution and tumor characteristics together suggest that H3.3 mutations arise at different developmental timepoints as well as from independent niches and cellular precursors within the brain.
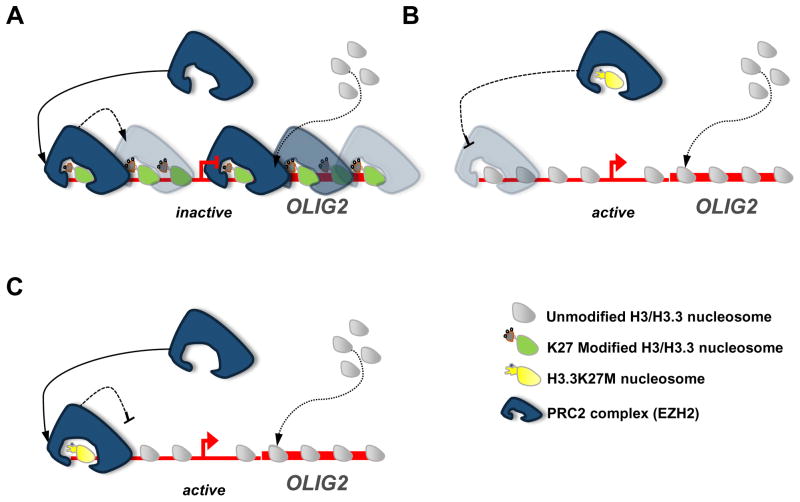
Mutant H3.3K27M can interact with PRC2 to alter transcription in a number of possible scenarios using a representative gene that is dysregulated in K27M gliomas (OLIG2). (A). “Wild type.” In this context, H3.3K27M has not been incorporated into the chromatin of OLIG2 (no mutant protein present) and thus does not inhibit PRC2 function. PRC2 is able to trimethylate H3K27, spreading H3K27me3 marks throughout the promoter region and gene body, effectively repressing transcription. (B). “Sequestered.” H3.3K27M peptide mimics K27 methylation and allosterically inhibits EZH2 methyltransferase function outside of chromatin. Free H3.3K27M in the nucleoplasm may bind to EZH2 through interaction with the active site, sequestering it in the nucleoplasm. Such binding lessens the likelihood of EZH2 binding and silencing its genomic targets. (C). “Trapped.” In this scenario, H3.3K27M has been incorporated into the promoter/transcriptional start site region. However, binding of EZH2 to H3.3K27M blocks the active site and interferes with methyltransferase activity of EZH2 on the same and nearby nucleosomes. Thus, this region is not properly silenced and does not recruit additional silencing factors to spread H3K27me3.
References
-
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9:1191–1200. - PubMed
-
- Akhmanova AS, Bindels PC, Xu J, Miedema K, Kremer H, Hennig W. Structure and expression of histone H3.3 genes in Drosophila melanogaster and Drosophila hydei. Genome. 1995;38:586–600. - PubMed
-
- Albig W, Bramlage B, Gruber K, Klobeck HG, Kunz J, Doenecke D. The human replacement histone H3.3B gene (H3F3B) Genomics. 1995;30:264–272. - PubMed
-
- Attieh Y, Geng QR, Dinardo CD, Zheng H, Jia Y, Fang ZH, Ganan-Gomez I, Yang H, Wei Y, Kantarjian H, et al. Low frequency of H3.3 mutations and upregulated DAXX expression in MDS. Blood. 2013;121:4009–4011. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
