

Human pluripotent stem cell models of autism spectrum disorder: emerging frontiers, opportunities, and challenges towards neuronal networks in a dish
- PMID: 24232378
- PMCID: PMC3932166
- DOI: 10.1007/s00213-013-3332-1
Human pluripotent stem cell models of autism spectrum disorder: emerging frontiers, opportunities, and challenges towards neuronal networks in a dish
Abstract
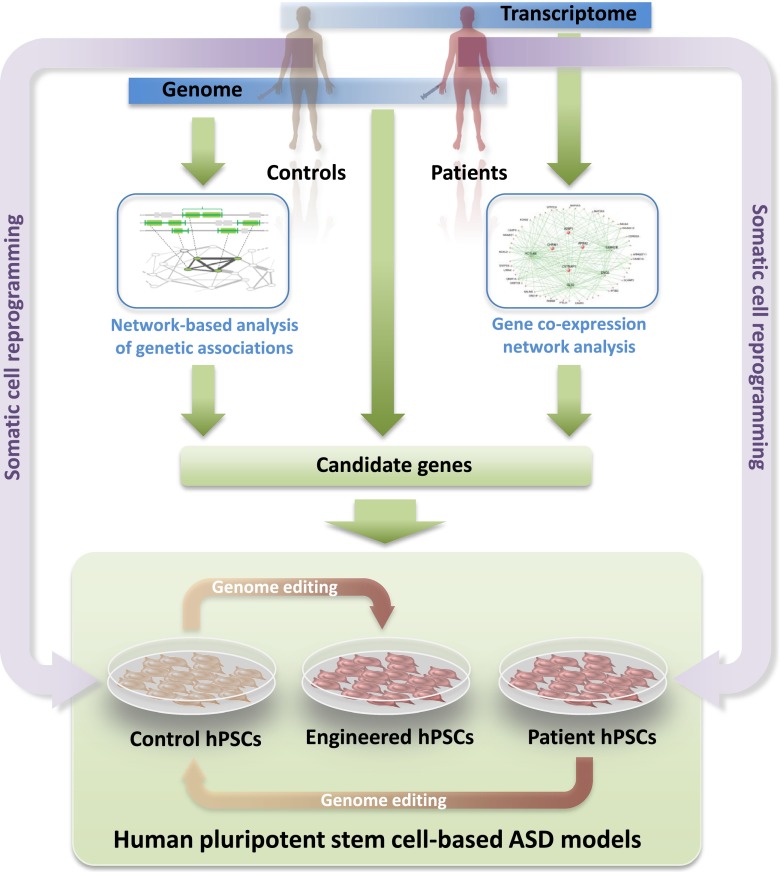
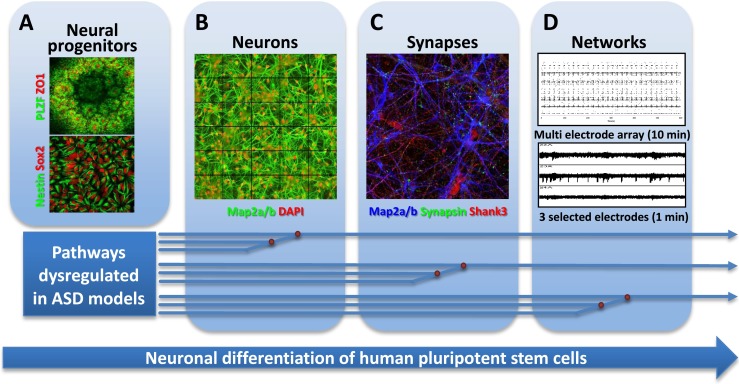
Autism spectrum disorder (ASD) is characterized by deficits in language development and social cognition and the manifestation of repetitive and restrictive behaviors. Despite recent major advances, our understanding of the pathophysiological mechanisms leading to ASD is limited. Although most ASD cases have unknown genetic underpinnings, animal and human cellular models of several rare, genetically defined syndromic forms of ASD have provided evidence for shared pathophysiological mechanisms that may extend to idiopathic cases. Here, we review our current knowledge of the genetic basis and molecular etiology of ASD and highlight how human pluripotent stem cell-based disease models have the potential to advance our understanding of molecular dysfunction. We summarize landmark studies in which neuronal cell populations generated from human embryonic stem cells and patient-derived induced pluripotent stem cells have served to model disease mechanisms, and we discuss recent technological advances that may ultimately allow in vitro modeling of specific human neuronal circuitry dysfunction in ASD. We propose that these advances now offer an unprecedented opportunity to help better understand ASD pathophysiology. This should ultimately enable the development of cellular models for ASD, allowing drug screening and the identification of molecular biomarkers for patient stratification.
Figures
References
-
- Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, Edel M, Boue S, Izpisua Belmonte JC. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. - PubMed
-
- Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–145. - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. - PubMed
-
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201. - PubMed
-
- Armstrong DD, Dunn K, Antalffy B. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J Neuropathol Exp Neurol. 1998;57:1013–1017. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
