

Physiological and environmental control of yeast prions
- PMID: 24236638
- PMCID: PMC3951574
- DOI: 10.1111/1574-6976.12053
Physiological and environmental control of yeast prions
Abstract
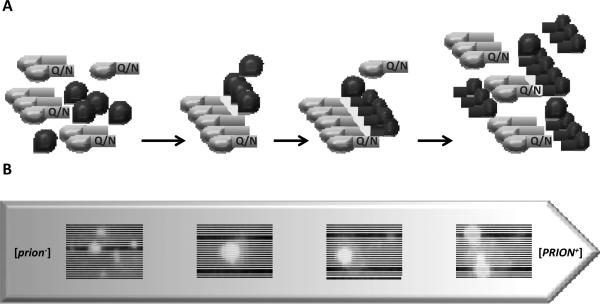
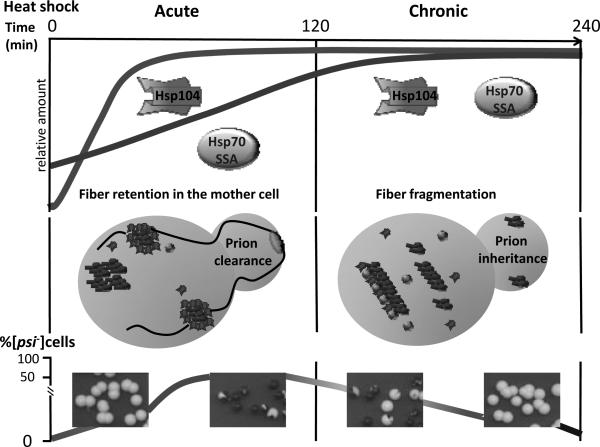
Prions are self-perpetuating protein isoforms that cause fatal and incurable neurodegenerative disease in mammals. Recent evidence indicates that a majority of human proteins involved in amyloid and neural inclusion disorders possess at least some prion properties. In lower eukaryotes, such as yeast, prions act as epigenetic elements, which increase phenotypic diversity by altering a range of cellular processes. While some yeast prions are clearly pathogenic, it is also postulated that prion formation could be beneficial in variable environmental conditions. Yeast and mammalian prions have similar molecular properties. Crucial cellular factors and conditions influencing prion formation and propagation were uncovered in the yeast models. Stress-related chaperones, protein quality control deposits, degradation pathways, and cytoskeletal networks control prion formation and propagation in yeast. Environmental stresses trigger prion formation and loss, supposedly acting via influencing intracellular concentrations of the prion-inducing proteins, and/or by localizing prionogenic proteins to the prion induction sites via heterologous ancillary helpers. Physiological and environmental modulation of yeast prions points to new opportunities for pharmacological intervention and/or prophylactic measures targeting general cellular systems rather than the properties of individual amyloids and prions.
Keywords: amyloid; chaperone; cytoskeleton; heat shock; quality control; ubiquitin.
© 2013 Federation of European Microbiological Societies. Published by John Wiley & Sons Ltd. All rights reserved.
Figures


References
-
- Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299:1751–1753. - PubMed
-
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–790. - PubMed
-
- Aguzzi A, O'Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–248. - PubMed
-
- Allen KD, Chernova TA, Tennant EP, Wilkinson KD, Chernoff YO. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J Biol Chem. 2007;282:3004–3013. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
