

The neuroimmune basis of fatigue
- PMID: 24239063
- PMCID: PMC3889707
- DOI: 10.1016/j.tins.2013.10.003
The neuroimmune basis of fatigue
Abstract
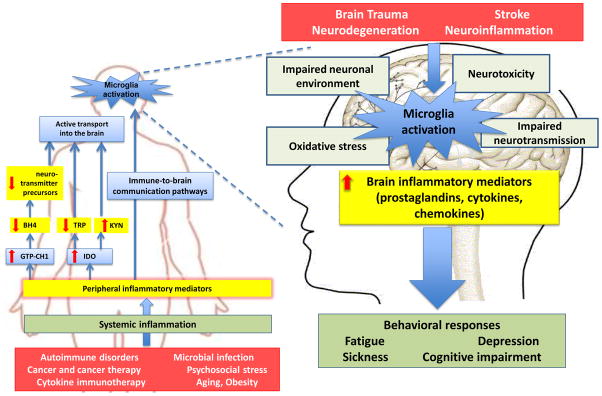
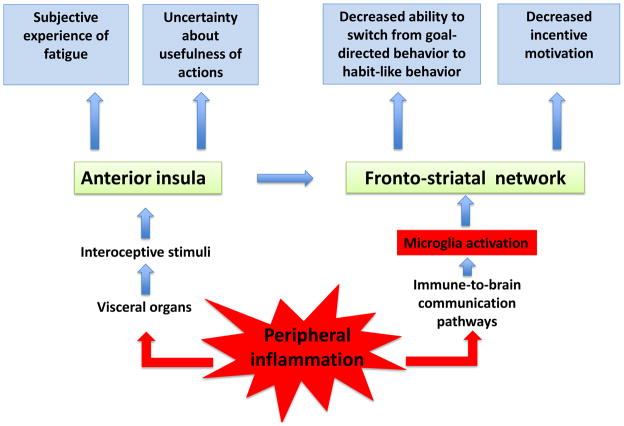
The exact nature and pathophysiology of fatigue remain largely elusive despite its high prevalence in physically ill patients. Studies on the relationship between the immune system and the central nervous system provide a new perspective on the mechanisms of fatigue. Inflammatory mediators that are released by activated innate immune cells at the periphery and in the central nervous system alter the metabolism and activity of neurotransmitters, generate neurotoxic compounds, decrease neurotrophic factors, and profoundly disturb the neuronal environment. The resulting alterations in fronto-striatal networks together with the activation of insula by inflammatory interoceptive stimuli underlie the many dimensions of fatigue including reduced incentive motivation, decreased behavioral flexibility, uncertainty about usefulness of actions, and awareness of fatigue.
Published by Elsevier Ltd.
Conflict of interest statement
R. Dantzer works as a consultant for Ironwood Pharma, Cambridge, MA
Figures
References
-
- Chaudhuri A, Behan PO. Fatigue and basal ganglia. J Neurol Sci. 2000;179(S 1–2):34–42. - PubMed
-
- Kroenke K, Price RK. Symptoms in the community. Prevalence, classification, and psychiatric comorbidity. Arch Intern Med. 1993;153(21):2474–80. - PubMed
-
- van’t Leven M, Zielhuis GA, van der Meer JW, Verbeek AL, Bleijenberg G. Fatigue and chronic fatigue syndrome-like complaints in the general population. Eur J Public Health. 2010;20(3):251–7. - PubMed
-
- Kroenke K, Stump T, Clark DO, Callahan CM, McDonald CJ. Symptoms in hospitalized patients: outcome and satisfaction with care. Am J Med. 1999;107(5):425–31. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
