

Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans
- PMID: 24239381
- PMCID: PMC3852926
- DOI: 10.1016/j.ajhg.2013.10.011
Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans
Abstract
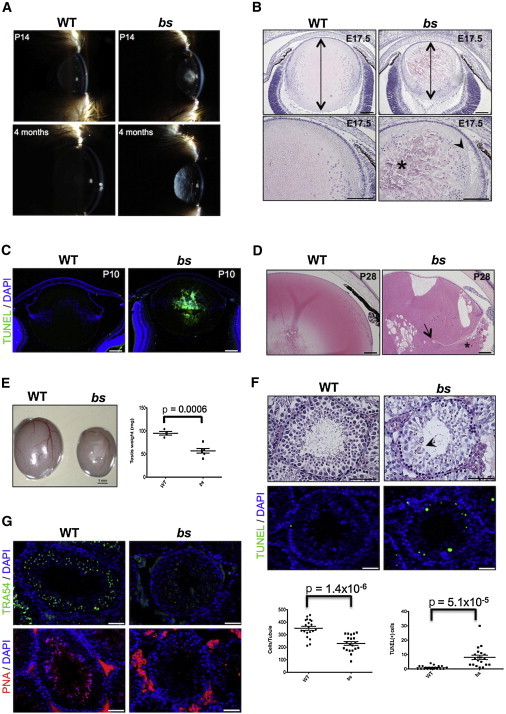
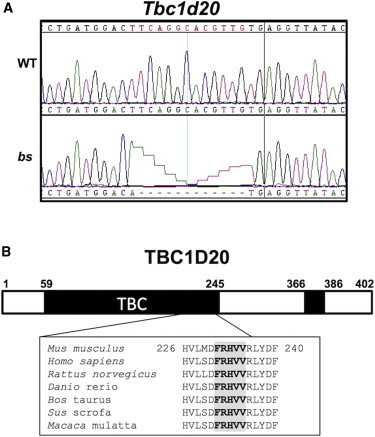
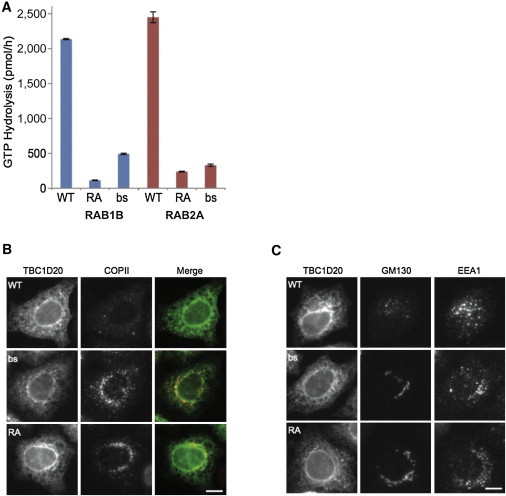
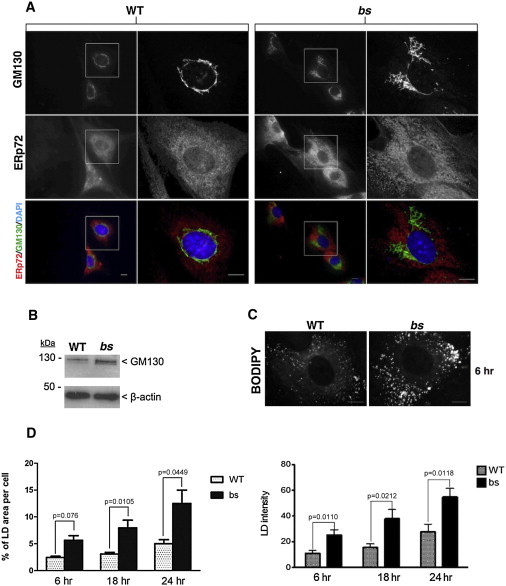
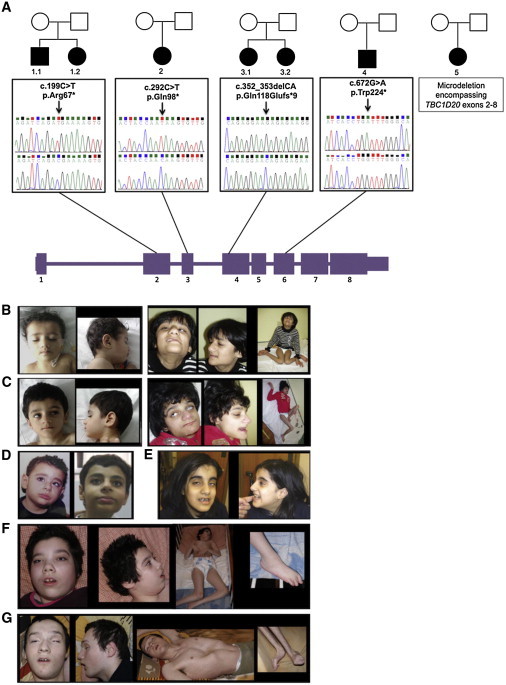
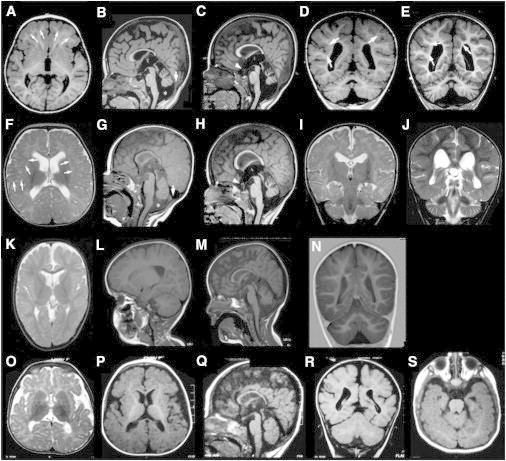
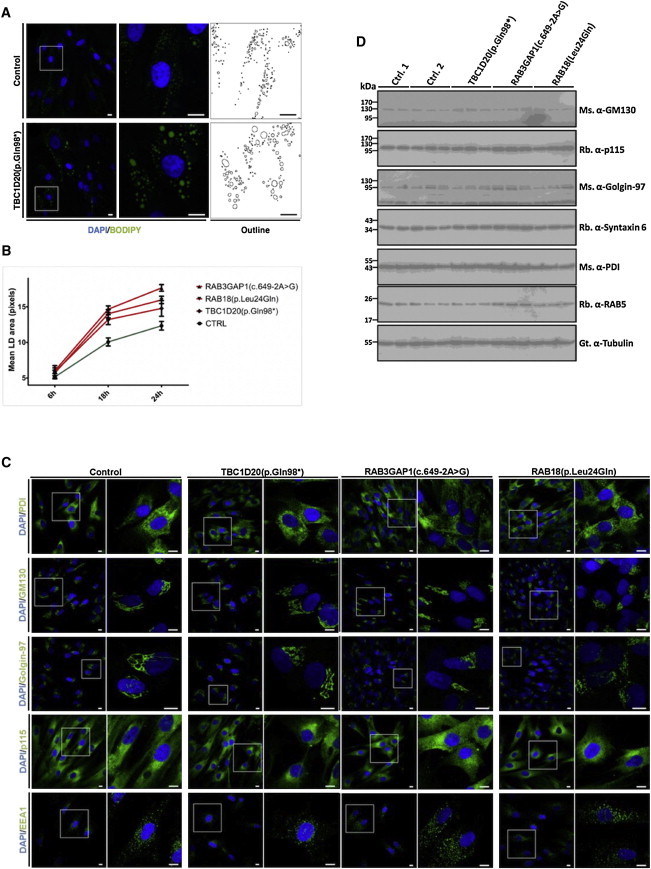
blind sterile (bs) is a spontaneous autosomal-recessive mouse mutation discovered more than 30 years ago. Phenotypically, bs mice exhibit nuclear cataracts and male infertility; genetic analyses assigned the bs locus to mouse chromosome 2. In this study, we first positionally cloned the bs locus and identified a putative causative mutation in the Tbc1d20 gene. Functional analysis established the mouse TBC1D20 protein as a GTPase-activating protein (GAP) for RAB1 and RAB2, and bs as a TBC1D20 loss-of-function mutation. Evaluation of bs mouse embryonic fibroblasts (mEFs) identified enlarged Golgi morphology and aberrant lipid droplet (LD) formation. Based on the function of TBC1D20 as a RABGAP and the bs cataract and testicular phenotypes, we hypothesized that mutations in TBC1D20 may contribute to Warburg micro syndrome (WARBM); WARBM constitutes a spectrum of disorders characterized by eye, brain, and endocrine abnormalities caused by mutations in RAB3GAP1, RAB3GAP2, and RAB18. Sequence analysis of a cohort of 77 families affected by WARBM identified five distinct TBC1D20 loss-of-function mutations, thereby establishing these mutations as causative of WARBM. Evaluation of human fibroblasts deficient in TBC1D20 function identified aberrant LDs similar to those identified in the bs mEFs. Additionally, our results show that human fibroblasts deficient in RAB18 and RAB3GAP1 function also exhibit aberrant LD formation. These findings collectively indicate that a defect in LD formation/metabolism may be a common cellular abnormality associated with WARBM, although it remains unclear whether abnormalities in LD metabolism are contributing to WARBM disease pathology.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Warburg M., Sjö O., Fledelius H.C., Pedersen S.A. Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am. J. Dis. Child. 1993;147:1309–1312. - PubMed
-
- Aligianis I.A., Johnson C.A., Gissen P., Chen D., Hampshire D., Hoffmann K., Maina E.N., Morgan N.V., Tee L., Morton J. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 2005;37:221–223. - PubMed
-
- Abdel-Salam G.M., Hassan N.A., Kayed H.F., Aligianis I.A. Phenotypic variability in Micro syndrome: report of new cases. Genet. Couns. 2007;18:423–435. - PubMed
-
- Morris-Rosendahl D.J., Segel R., Born A.P., Conrad C., Loeys B., Brooks S.S., Müller L., Zeschnigk C., Botti C., Rabinowitz R. New RAB3GAP1 mutations in patients with Warburg Micro Syndrome from different ethnic backgrounds and a possible founder effect in the Danish. Eur. J. Hum. Genet. 2010;18:1100–1106. - PMC - PubMed
-
- Handley M.T., Morris-Rosendahl D.J., Brown S., Macdonald F., Hardy C., Bem D., Carpanini S.M., Borck G., Martorell L., Izzi C. Mutation spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype-phenotype correlations in warburg micro syndrome and Martsolf syndrome. Hum. Mutat. 2013;34:686–696. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
