

PAIRpred: partner-specific prediction of interacting residues from sequence and structure
- PMID: 24243399
- PMCID: PMC4329725
- DOI: 10.1002/prot.24479
PAIRpred: partner-specific prediction of interacting residues from sequence and structure
Abstract
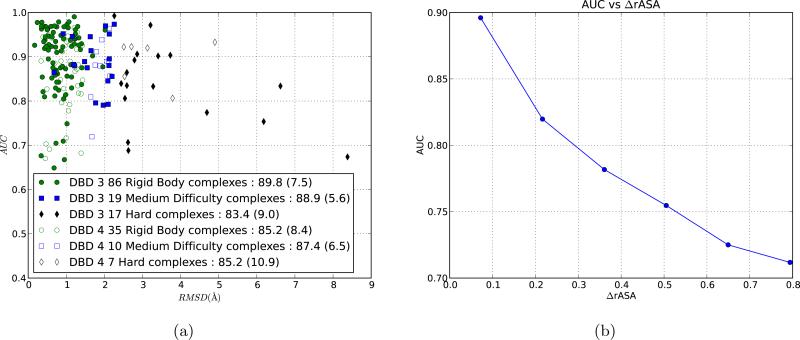
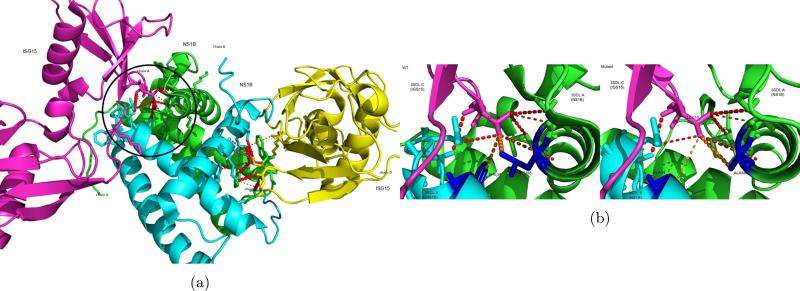
We present a novel partner-specific protein-protein interaction site prediction method called PAIRpred. Unlike most existing machine learning binding site prediction methods, PAIRpred uses information from both proteins in a protein complex to predict pairs of interacting residues from the two proteins. PAIRpred captures sequence and structure information about residue pairs through pairwise kernels that are used for training a support vector machine classifier. As a result, PAIRpred presents a more detailed model of protein binding, and offers state of the art accuracy in predicting binding sites at the protein level as well as inter-protein residue contacts at the complex level. We demonstrate PAIRpred's performance on Docking Benchmark 4.0 and recent CAPRI targets. We present a detailed performance analysis outlining the contribution of different sequence and structure features, together with a comparison to a variety of existing interface prediction techniques. We have also studied the impact of binding-associated conformational change on prediction accuracy and found PAIRpred to be more robust to such structural changes than existing schemes. As an illustration of the potential applications of PAIRpred, we provide a case study in which PAIRpred is used to analyze the nature and specificity of the interface in the interaction of human ISG15 protein with NS1 protein from influenza A virus. Python code for PAIRpred is available at http://combi.cs.colostate.edu/supplements/pairpred/.
Keywords: protein binding site prediction; protein interface prediction.
© 2013 Wiley Periodicals, Inc.
Figures
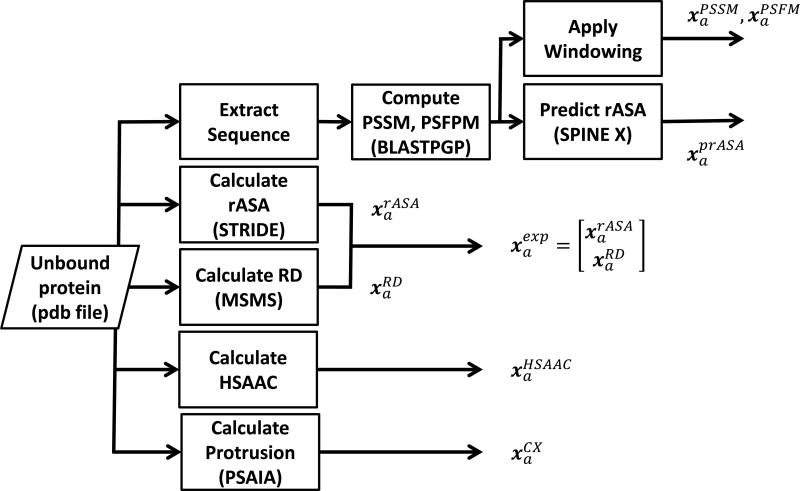

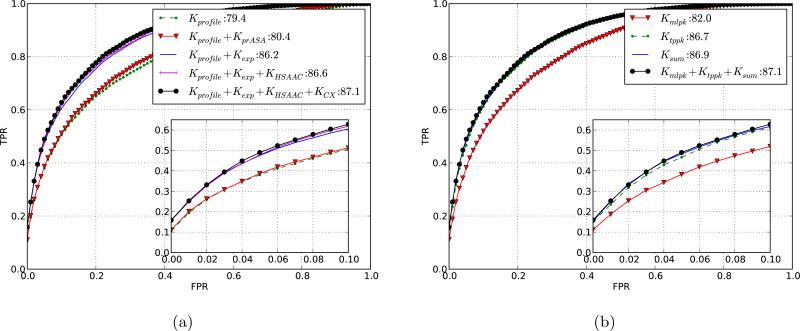
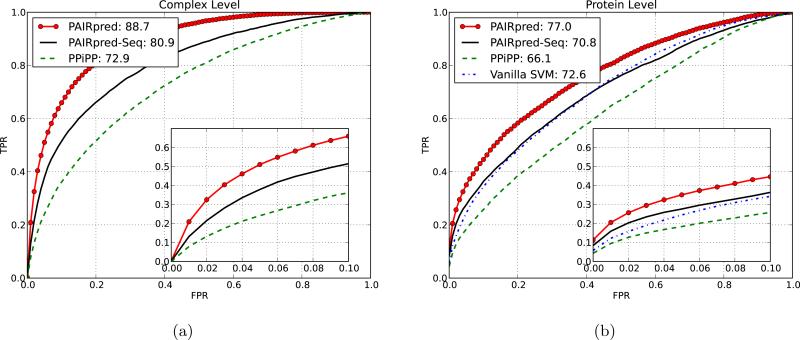
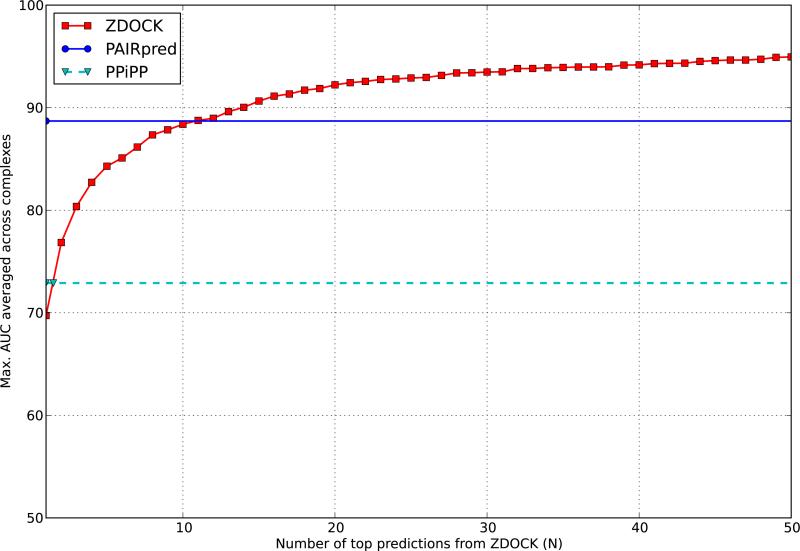
References
-
- Ezkurdia I, Bartoli L, Fariselli P, Casadio R, Valencia A, Tress ML. Progress and challenges in predicting protein-protein interaction sites. Briefings in Bioinformatics. 2009 May;10:233–246. - PubMed
-
- Keskin O, Gursoy A, Ma B, Nussinov R. Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chemical reviews. 2008 Apr.108:1225–1244. PMID: 18355092. - PubMed
-
- Gunasekaran K, Nussinov R. How different are structurally flexible and rigid binding sites? sequence and structural features discriminating proteins that do and do not undergo conformational change upon ligand binding. Journal of Molecular Biology. 2007 Jan.365:257–273. - PubMed
-
- Wass MN, David A, Sternberg MJ. Challenges for the prediction of macro-molecular interactions. Current Opinion in Structural Biology. 2011 Jun;21:382–390. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
