

Catalysis of protein folding by chaperones accelerates evolutionary dynamics in adapting cell populations
- PMID: 24244114
- PMCID: PMC3820506
- DOI: 10.1371/journal.pcbi.1003269
Catalysis of protein folding by chaperones accelerates evolutionary dynamics in adapting cell populations
Abstract
Although molecular chaperones are essential components of protein homeostatic machinery, their mechanism of action and impact on adaptation and evolutionary dynamics remain controversial. Here we developed a physics-based ab initio multi-scale model of a living cell for population dynamics simulations to elucidate the effect of chaperones on adaptive evolution. The 6-loci genomes of model cells encode model proteins, whose folding and interactions in cellular milieu can be evaluated exactly from their genome sequences. A genotype-phenotype relationship that is based on a simple yet non-trivially postulated protein-protein interaction (PPI) network determines the cell division rate. Model proteins can exist in native and molten globule states and participate in functional and all possible promiscuous non-functional PPIs. We find that an active chaperone mechanism, whereby chaperones directly catalyze protein folding, has a significant impact on the cellular fitness and the rate of evolutionary dynamics, while passive chaperones, which just maintain misfolded proteins in soluble complexes have a negligible effect on the fitness. We find that by partially releasing the constraint on protein stability, active chaperones promote a deeper exploration of sequence space to strengthen functional PPIs, and diminish the non-functional PPIs. A key experimentally testable prediction emerging from our analysis is that down-regulation of chaperones that catalyze protein folding significantly slows down the adaptation dynamics.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
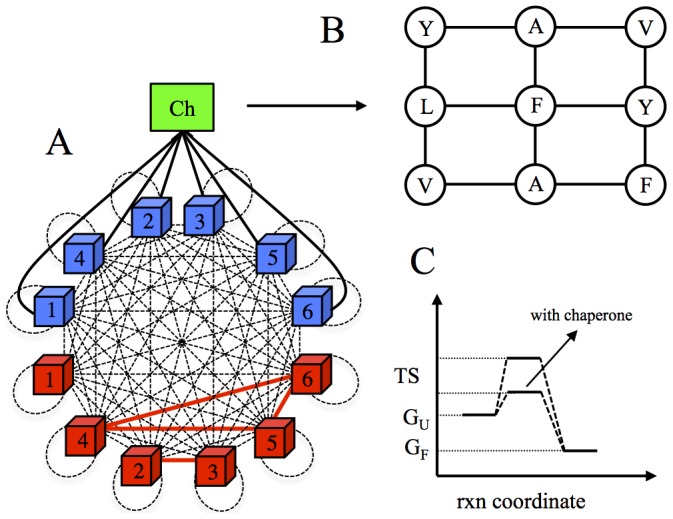
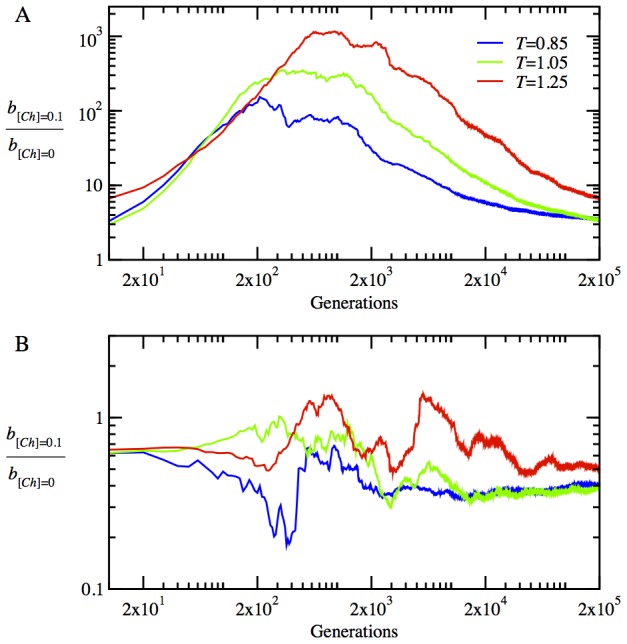
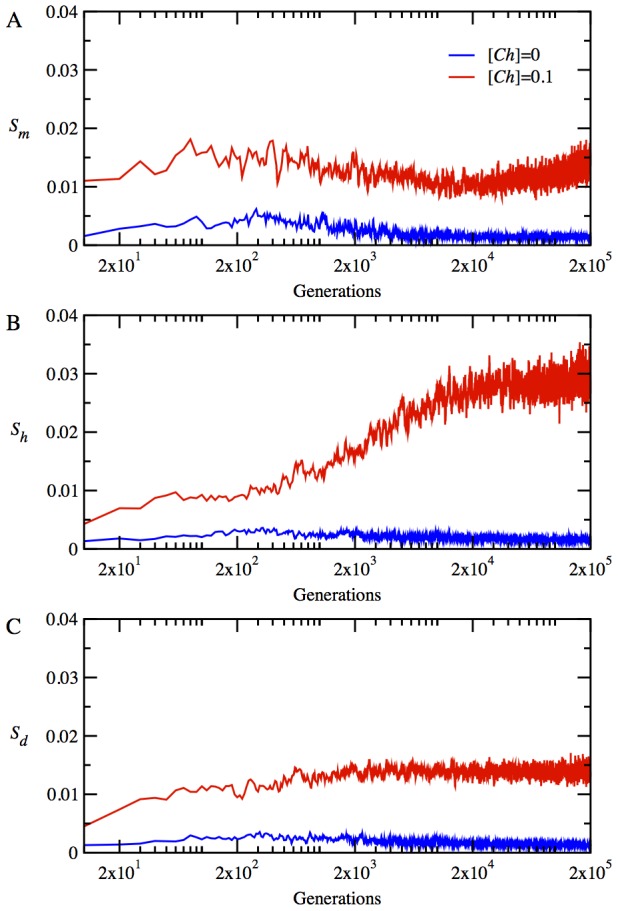
 for monomeric proteins. (B) The time evolution of mean stability,
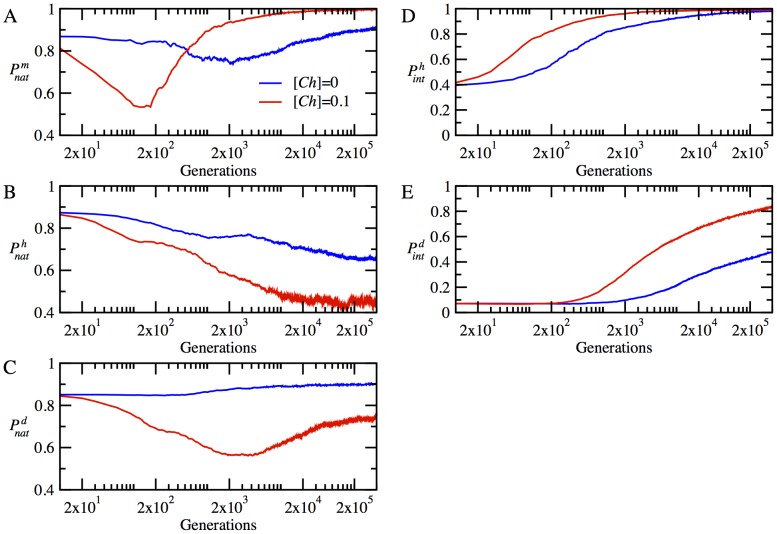
for monomeric proteins. (B) The time evolution of mean stability,  for heterodimer proteins. (C) The time evolution of mean stability,
for heterodimer proteins. (C) The time evolution of mean stability,  for date triangle proteins. (D) The time evolution of interaction probability,
for date triangle proteins. (D) The time evolution of interaction probability,  for the heterodimer complexes. (E) The time evolution of mean interaction probability,
for the heterodimer complexes. (E) The time evolution of mean interaction probability,  for the date triangle complexes.
for the date triangle complexes.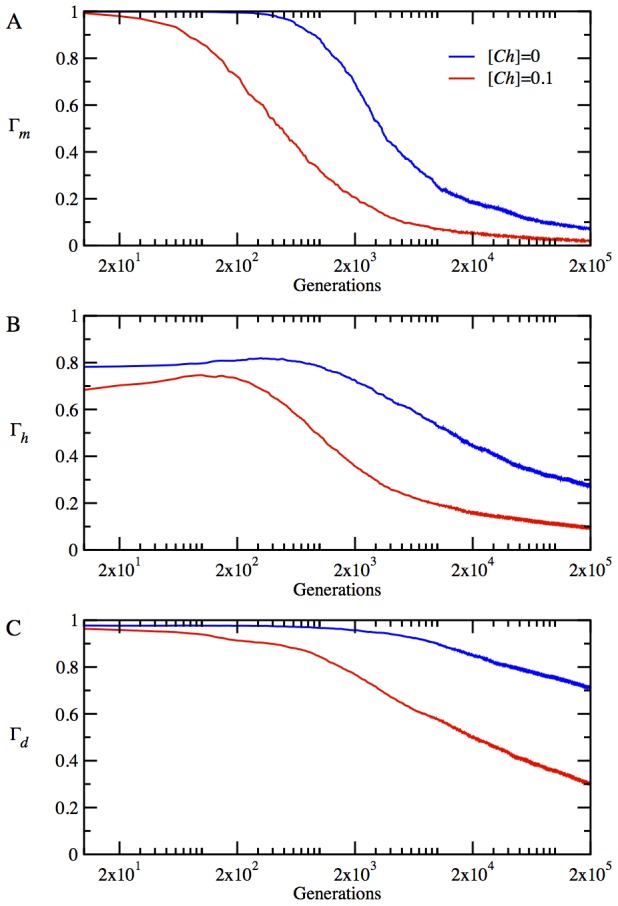
 (B) average NF-PPI for heterodimers
(B) average NF-PPI for heterodimers  and (C) NF-PPI for date triangles
and (C) NF-PPI for date triangles  where
where 


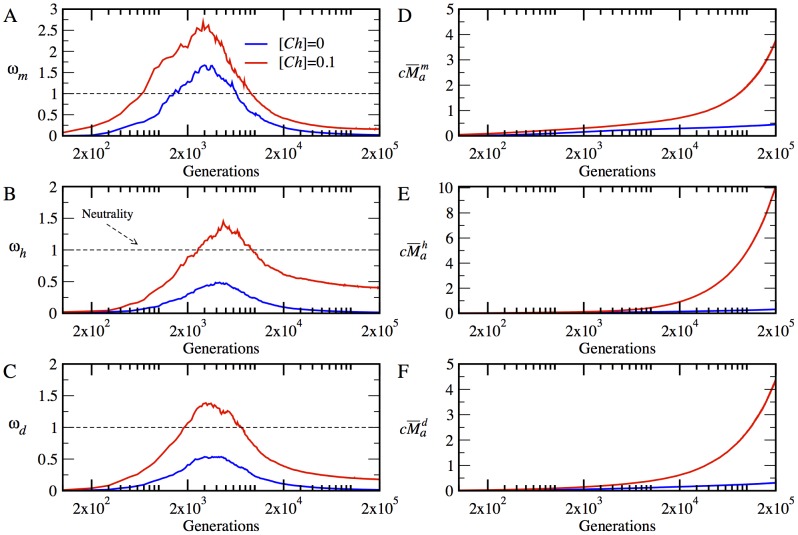
 represents the neutral evolution. The time evolution of
represents the neutral evolution. The time evolution of  is plotted in (A) for the monomer
is plotted in (A) for the monomer  , in (B) for the heterodimer
, in (B) for the heterodimer  , and in (C), for the date triangle
, and in (C), for the date triangle  . The time evolution of
. The time evolution of  is plotted in (D) for the monomer
is plotted in (D) for the monomer  , in (E) for the heterodimer
, in (E) for the heterodimer  , and in (F) for the date triangle
, and in (F) for the date triangle  .
.
 , in (B) for the heterodimer
, in (B) for the heterodimer  , and in (C) for date triangle proteins
, and in (C) for date triangle proteins  .
.Similar articles
-
Is catalytic activity of chaperones a selectable trait for the emergence of heat shock response?Biophys J. 2015 Jan 20;108(2):438-48. doi: 10.1016/j.bpj.2014.11.3468. Biophys J. 2015. PMID: 25606691 Free PMC article.
-
Protein-Protein Interactions in the Molecular Chaperone Network.Acc Chem Res. 2018 Apr 17;51(4):940-949. doi: 10.1021/acs.accounts.8b00036. Epub 2018 Apr 3. Acc Chem Res. 2018. PMID: 29613769 Free PMC article. Review.
-
Binding and folding: in search of intramolecular chaperone-like building block fragments.Protein Eng. 2000 Sep;13(9):617-27. doi: 10.1093/protein/13.9.617. Protein Eng. 2000. PMID: 11054456
-
Molecular Chaperones Accelerate the Evolution of Their Protein Clients in Yeast.Genome Biol Evol. 2019 Aug 1;11(8):2360-2375. doi: 10.1093/gbe/evz147. Genome Biol Evol. 2019. PMID: 31297528 Free PMC article.
-
Cumulative impact of chaperone-mediated folding on genome evolution.Biochemistry. 2012 Dec 18;51(50):9941-53. doi: 10.1021/bi3013643. Epub 2012 Dec 10. Biochemistry. 2012. PMID: 23167595 Review.
Cited by
-
Could Small Heat Shock Protein HSP27 Be a First-Line Target for Preventing Protein Aggregation in Parkinson's Disease?Int J Mol Sci. 2021 Mar 16;22(6):3038. doi: 10.3390/ijms22063038. Int J Mol Sci. 2021. PMID: 33809767 Free PMC article. Review.
-
Viral Evolution Shaped by Host Proteostasis Networks.Annu Rev Virol. 2023 Sep 29;10(1):77-98. doi: 10.1146/annurev-virology-100220-112120. Epub 2023 Apr 18. Annu Rev Virol. 2023. PMID: 37071930 Free PMC article. Review.
-
The endoplasmic reticulum proteostasis network profoundly shapes the protein sequence space accessible to HIV envelope.PLoS Biol. 2022 Feb 18;20(2):e3001569. doi: 10.1371/journal.pbio.3001569. eCollection 2022 Feb. PLoS Biol. 2022. PMID: 35180219 Free PMC article.
-
The C-terminal tails of the bacterial chaperonin GroEL stimulate protein folding by directly altering the conformation of a substrate protein.J Biol Chem. 2014 Aug 15;289(33):23219-23232. doi: 10.1074/jbc.M114.577205. Epub 2014 Jun 25. J Biol Chem. 2014. PMID: 24970895 Free PMC article.
-
Is catalytic activity of chaperones a selectable trait for the emergence of heat shock response?Biophys J. 2015 Jan 20;108(2):438-48. doi: 10.1016/j.bpj.2014.11.3468. Biophys J. 2015. PMID: 25606691 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
