

Structural features of the regulatory ACT domain of phenylalanine hydroxylase
- PMID: 24244510
- PMCID: PMC3828330
- DOI: 10.1371/journal.pone.0079482
Structural features of the regulatory ACT domain of phenylalanine hydroxylase
Abstract
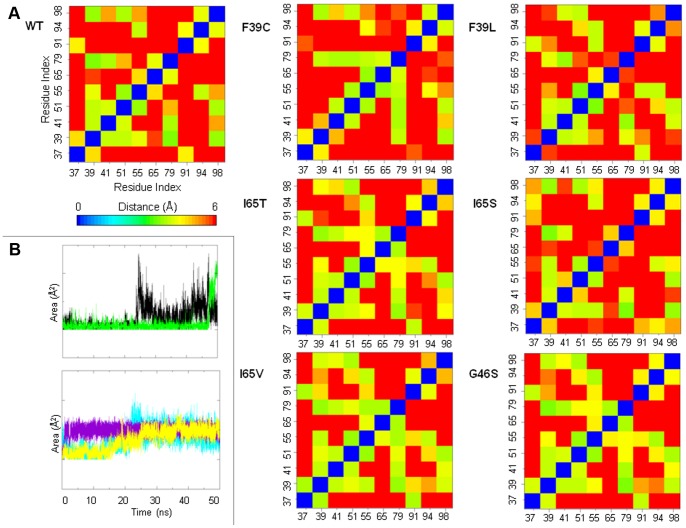
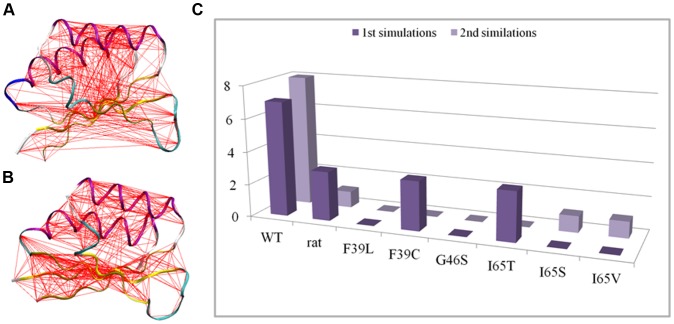
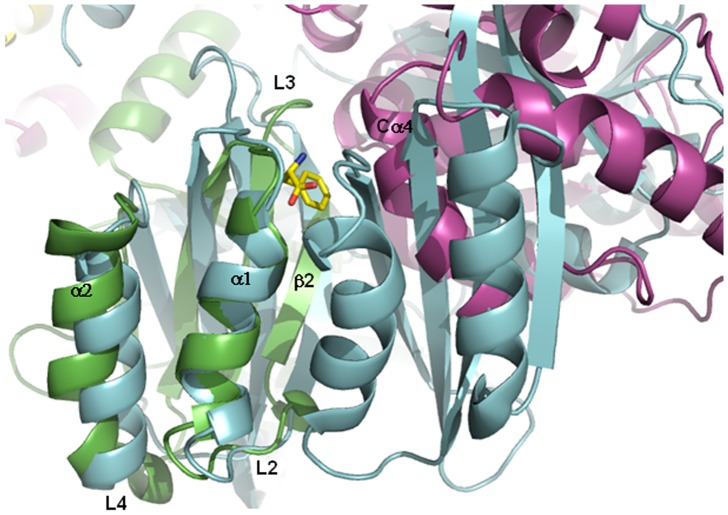
Phenylalanine hydroxylase (PAH) catalyzes the conversion of L-Phe to L-Tyr. Defects in PAH activity, caused by mutations in the human gene, result in the autosomal recessively inherited disease hyperphenylalaninemia. PAH activity is regulated by multiple factors, including phosphorylation and ligand binding. In particular, PAH displays positive cooperativity for L-Phe, which is proposed to bind the enzyme on an allosteric site in the N-terminal regulatory domain (RD), also classified as an ACT domain. This domain is found in several proteins and is able to bind amino acids. We used molecular dynamics simulations to obtain dynamical and structural insights into the isolated RD of PAH. Here we show that the principal motions involve conformational changes leading from an initial open to a final closed domain structure. The global intrinsic motions of the RD are correlated with exposure to solvent of a hydrophobic surface, which corresponds to the ligand binding-site of the ACT domain. Our results strongly suggest a relationship between the Phe-binding function and the overall dynamic behaviour of the enzyme. This relationship may be affected by structure-disturbing mutations. To elucidate the functional implications of the mutations, we investigated the structural effects on the dynamics of the human RD PAH induced by six missense hyperphenylalaninemia-causing mutations, namely p.G46S, p.F39C, p.F39L, p.I65S, p.I65T and p.I65V. These studies showed that the alterations in RD hydrophobic interactions induced by missense mutations could affect the functionality of the whole enzyme.
Conflict of interest statement
Figures

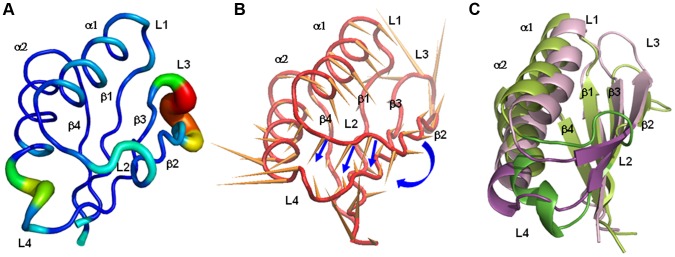
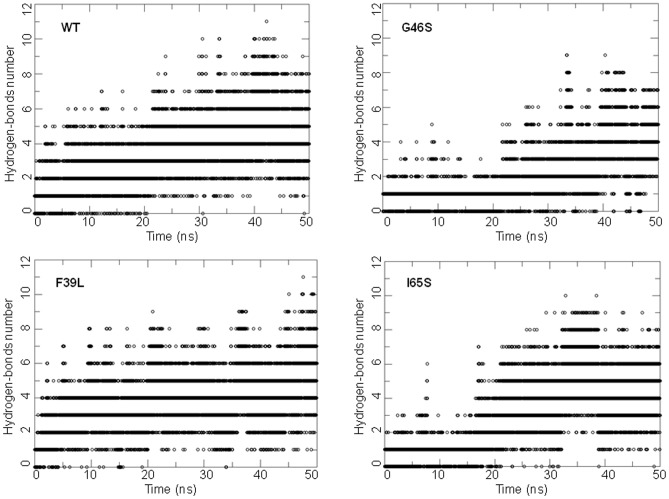
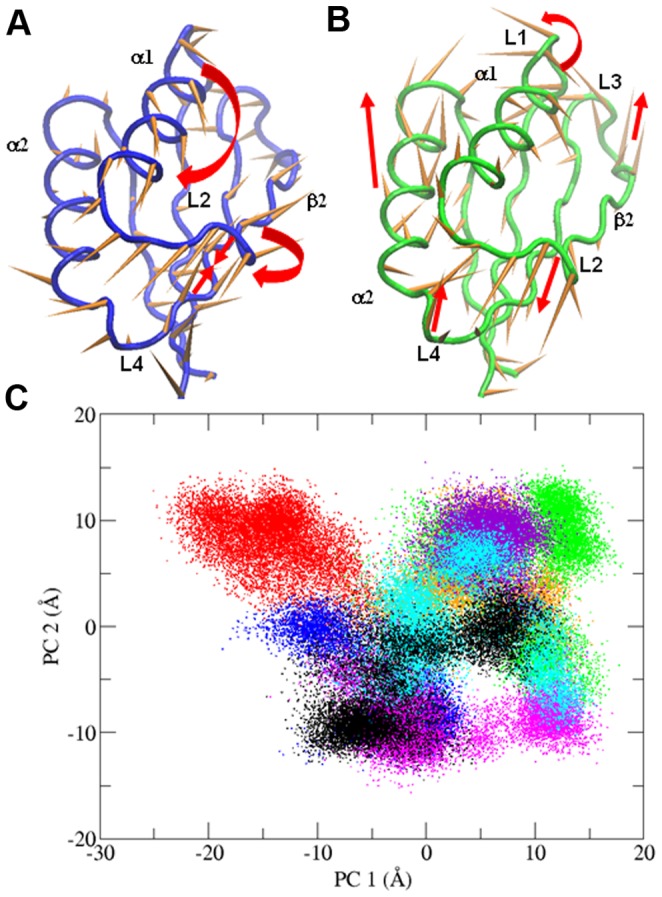
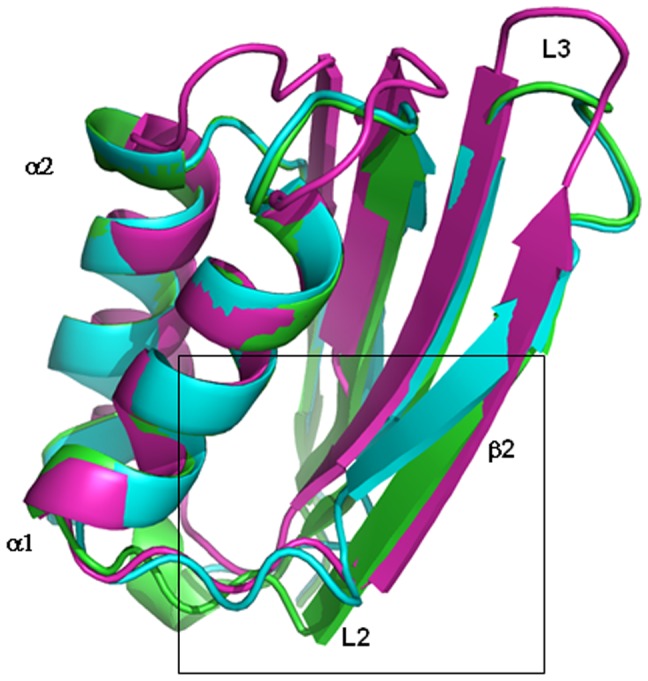


References
-
- Kaufman S (1993) The phenylalanine hydroxylating system. Adv Enzymol 70: 77–264. - PubMed
-
- Erlandsen H, Stevens RC (1999) The structural basis of phenylketonuria. Mol Genet Metab 68: 103–125. - PubMed
-
- Okano Y, Wang T, Eisensmith RC, Longhi R, Riva E, et al. (1991) Phenylketonuria missense mutations in the Mediterranean. Genomics 9: 96–103. - PubMed
-
- Svensson E, Iselius L, Hagenfeldt L (1994) Severity of mutation in the phenylalanine hydroxylase gene influences phenylalanine metabolism in phenylketonuria and hyperphenylalaninaemia heterozygotes. J Inherit Metab Dis 17: 215–222. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
