

Filamentous white matter prion protein deposition is a distinctive feature of multiple inherited prion diseases
- PMID: 24252267
- PMCID: PMC4046834
- DOI: 10.1186/2051-5960-1-8
Filamentous white matter prion protein deposition is a distinctive feature of multiple inherited prion diseases
Abstract
Background: Sporadic, inherited and acquired prion diseases show distinct histological patterns of abnormal prion protein (PrP) deposits. Many of the inherited prion diseases show striking histological patterns, which often associate with specific mutations. Most reports have focused on the pattern of PrP deposition in the cortical or cerebellar grey matter.
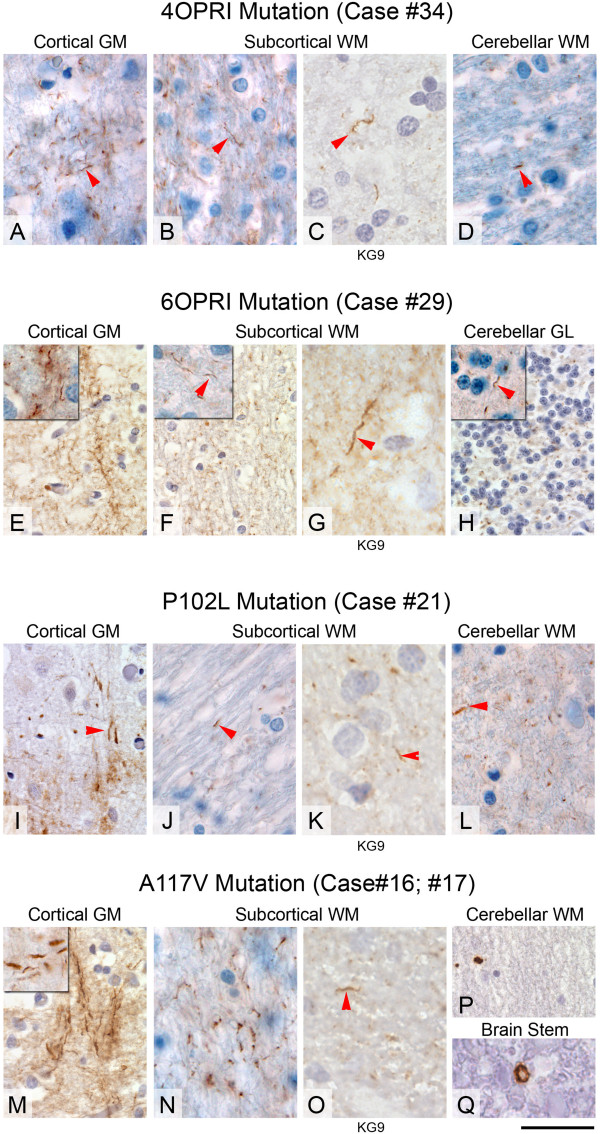
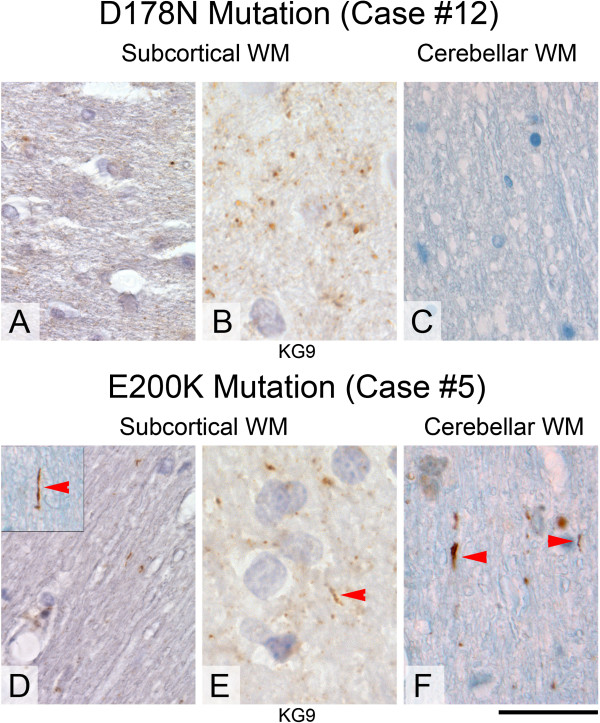
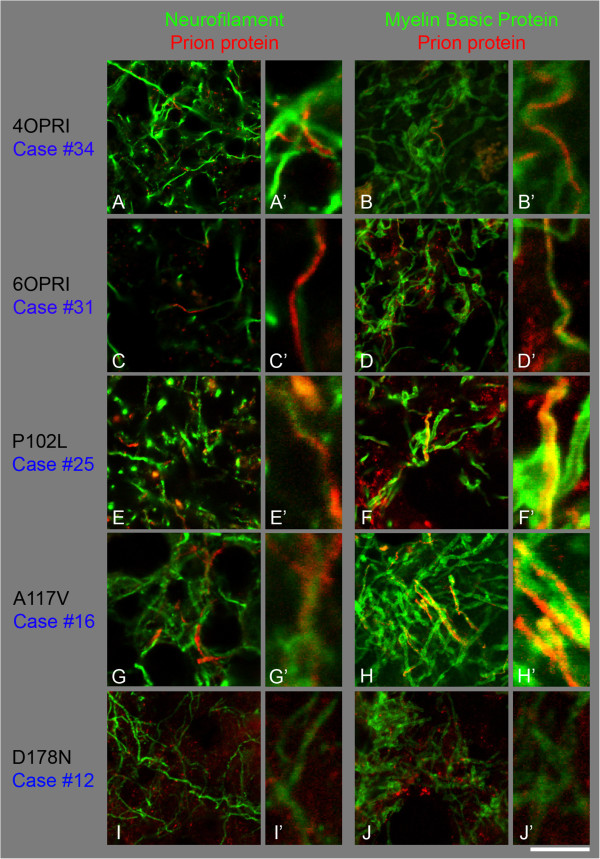
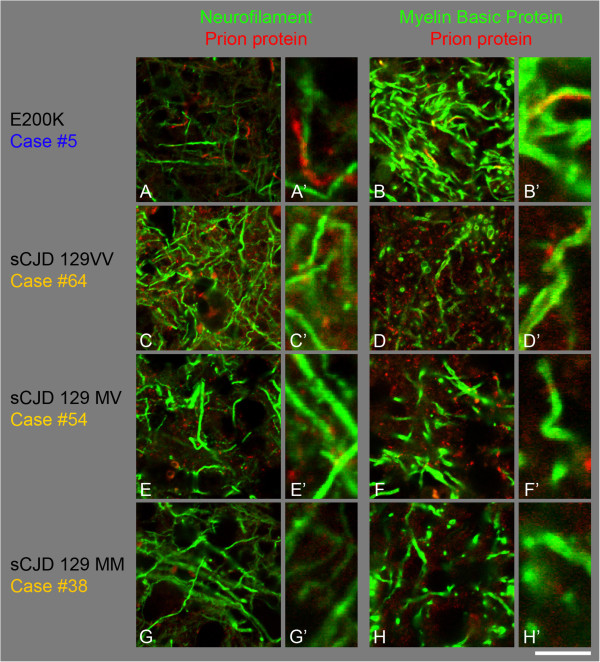
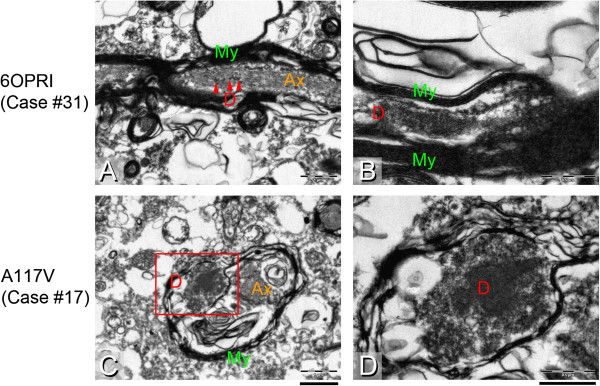
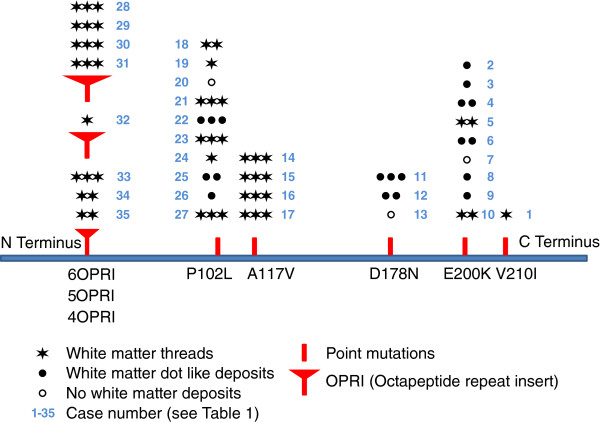
Results: We observed that the subcortical white matter in inherited prion diseases frequently contained filamentous depositions of abnormal PrP, and we have analysed by immunohistochemistry, immunofluorescence and electron microscopy 35 cases of inherited prion disease seen at the UK National Prion Clinic. We report here that filamentous PrP is abundantly deposited in myelinated fibres in inherited prion diseases, in particular in those with N-terminal mutations.
Conclusions: It is possible that the presence of filamentous PrP is related to the pathogenesis of inherited forms, which is different from those sporadic and acquired forms.
Figures
References
-
- Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases) Brain Pathol. 1995;5:459–466. doi: 10.1111/j.1750-3639.1995.tb00625.x. - DOI - PubMed
-
- Liberski PP, Sikorska B, Hauw JJ, Kopp N, Streichenberger N, Giraud P, Boellaard J, Budka H, Kovacs GG, Ironside J, Brown P. Ultrastructural characteristics (or evaluation) of Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies or prion diseases. Ultrastruct Pathol. 2010;34:351–361. doi: 10.3109/01913123.2010.491175. - DOI - PubMed
-
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. doi: 10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
