

Mitochondrial genome changes and neurodegenerative diseases
- PMID: 24252612
- PMCID: PMC4283582
- DOI: 10.1016/j.bbadis.2013.11.012
Mitochondrial genome changes and neurodegenerative diseases
Abstract
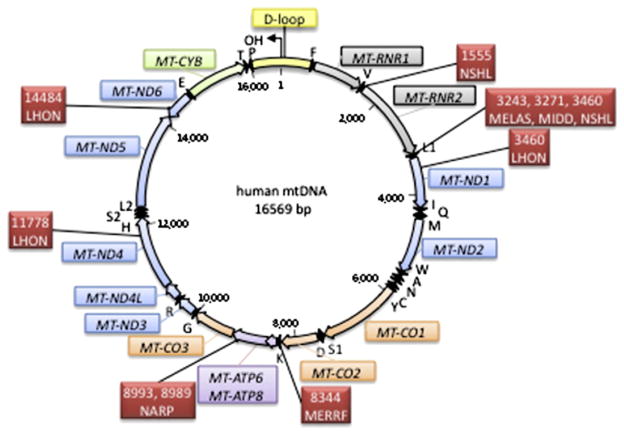
Mitochondria are essential organelles within the cell where most of the energy production occurs by the oxidative phosphorylation system (OXPHOS). Critical components of the OXPHOS are encoded by the mitochondrial DNA (mtDNA) and therefore, mutations involving this genome can be deleterious to the cell. Post-mitotic tissues, such as muscle and brain, are most sensitive to mtDNA changes, due to their high energy requirements and non-proliferative status. It has been proposed that mtDNA biological features and location make it vulnerable to mutations, which accumulate over time. However, although the role of mtDNA damage has been conclusively connected to neuronal impairment in mitochondrial diseases, its role in age-related neurodegenerative diseases remains speculative. Here we review the pathophysiology of mtDNA mutations leading to neurodegeneration and discuss the insights obtained by studying mouse models of mtDNA dysfunction.
Keywords: Encephalopathy; Mitochondrion; mtDNA.
Copyright © 2013. Published by Elsevier B.V.
Figures
Similar articles
-
Significance of Mitochondria DNA Mutations in Diseases.Adv Exp Med Biol. 2017;1038:219-230. doi: 10.1007/978-981-10-6674-0_15. Adv Exp Med Biol. 2017. PMID: 29178079 Review.
-
Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease.FEBS Lett. 2018 Mar;592(5):728-742. doi: 10.1002/1873-3468.12956. Epub 2018 Jan 9. FEBS Lett. 2018. PMID: 29281123 Free PMC article. Review.
-
The mitochondrial genome in human adaptive radiation and disease: on the road to therapeutics and performance enhancement.Gene. 2005 Jul 18;354:169-80. doi: 10.1016/j.gene.2005.05.001. Gene. 2005. PMID: 16024186 Review.
-
The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions.J Neurosci. 2011 Jul 6;31(27):9895-904. doi: 10.1523/JNEUROSCI.6223-10.2011. J Neurosci. 2011. PMID: 21734281 Free PMC article.
-
Mitochondria, oxidative stress and neurodegeneration.J Neurol Sci. 2012 Nov 15;322(1-2):254-62. doi: 10.1016/j.jns.2012.05.030. Epub 2012 Jun 4. J Neurol Sci. 2012. PMID: 22669122 Review.
Cited by
-
A Pilot Study of Bioenergetic Marker Relationships in Gulf War Illness: Phosphocreatine Recovery vs. Citric Acid Cycle Intermediates.Int J Environ Res Public Health. 2021 Feb 9;18(4):1635. doi: 10.3390/ijerph18041635. Int J Environ Res Public Health. 2021. PMID: 33572101 Free PMC article.
-
Association of mitochondrial variants and haplogroups identified by whole exome sequencing with Alzheimer's disease.Alzheimers Dement. 2022 Feb;18(2):294-306. doi: 10.1002/alz.12396. Epub 2021 Jun 20. Alzheimers Dement. 2022. PMID: 34152079 Free PMC article.
-
DNA2 mutation causing multisystemic disorder with impaired mitochondrial DNA maintenance.J Hum Genet. 2022 Dec;67(12):691-699. doi: 10.1038/s10038-022-01075-4. Epub 2022 Sep 5. J Hum Genet. 2022. PMID: 36064591 Free PMC article.
-
Mechanisms linking mtDNA damage and aging.Free Radic Biol Med. 2015 Aug;85:250-8. doi: 10.1016/j.freeradbiomed.2015.05.005. Epub 2015 May 13. Free Radic Biol Med. 2015. PMID: 25979659 Free PMC article. Review.
-
Mitochondrial Targeting in Neurodegeneration: A Heme Perspective.Pharmaceuticals (Basel). 2018 Sep 18;11(3):87. doi: 10.3390/ph11030087. Pharmaceuticals (Basel). 2018. PMID: 30231533 Free PMC article. Review.
References
-
- Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
