

Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer's disease cybrid cell
- PMID: 24252614
- PMCID: PMC3991235
- DOI: 10.1016/j.bbadis.2013.11.009
Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer's disease cybrid cell
Abstract
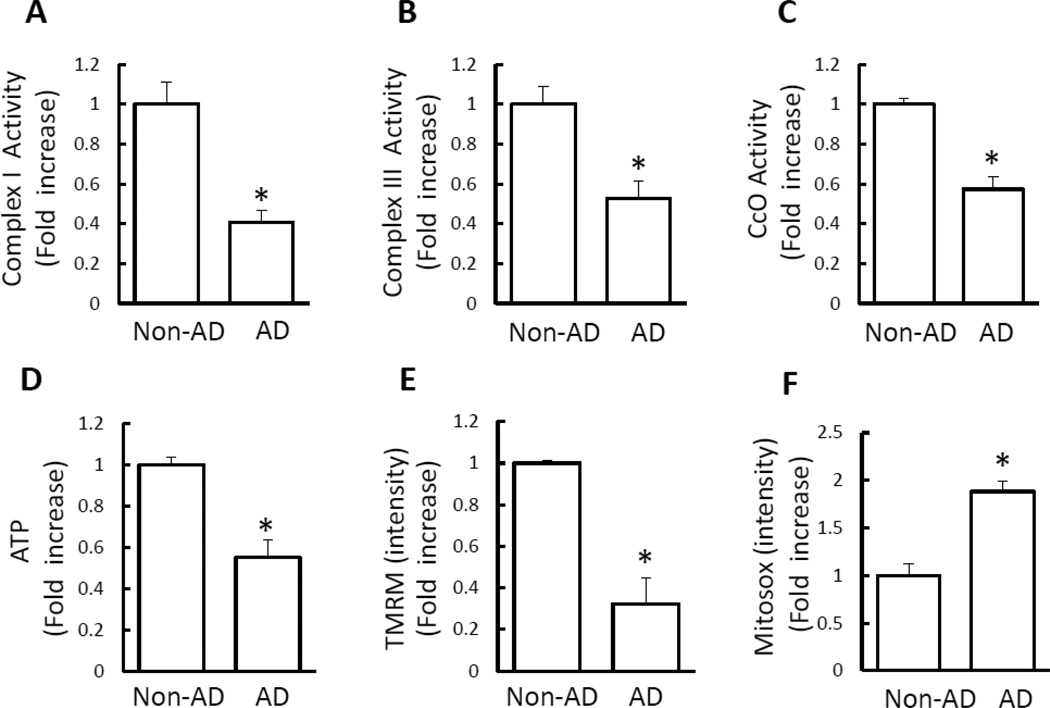
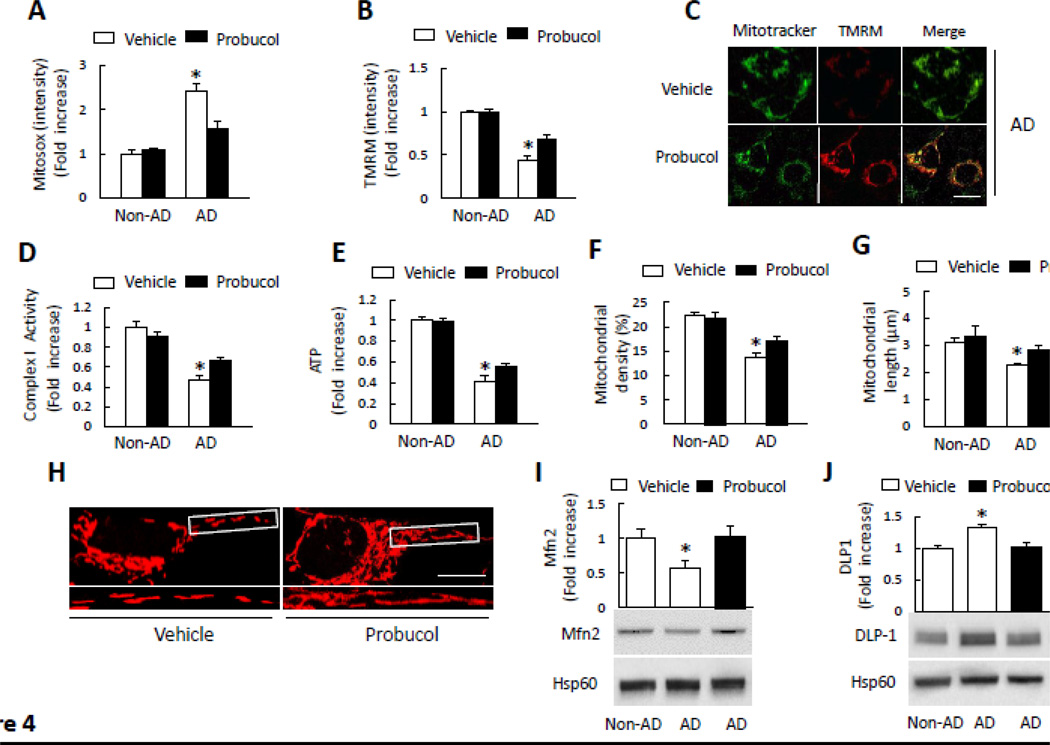
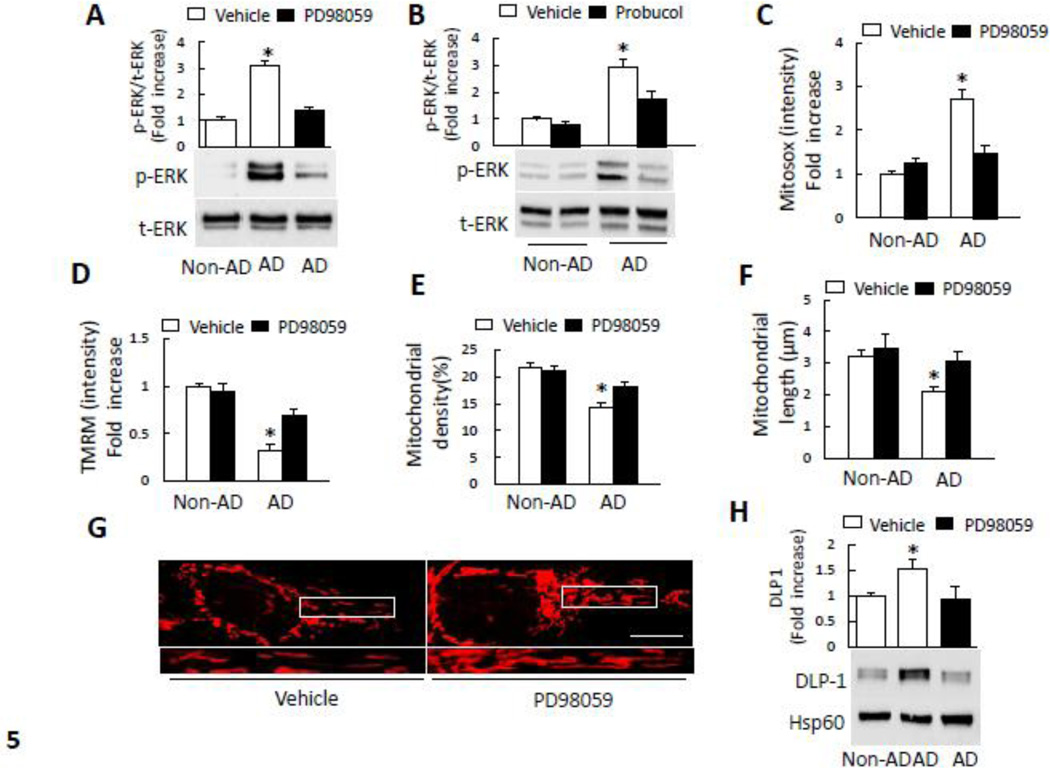
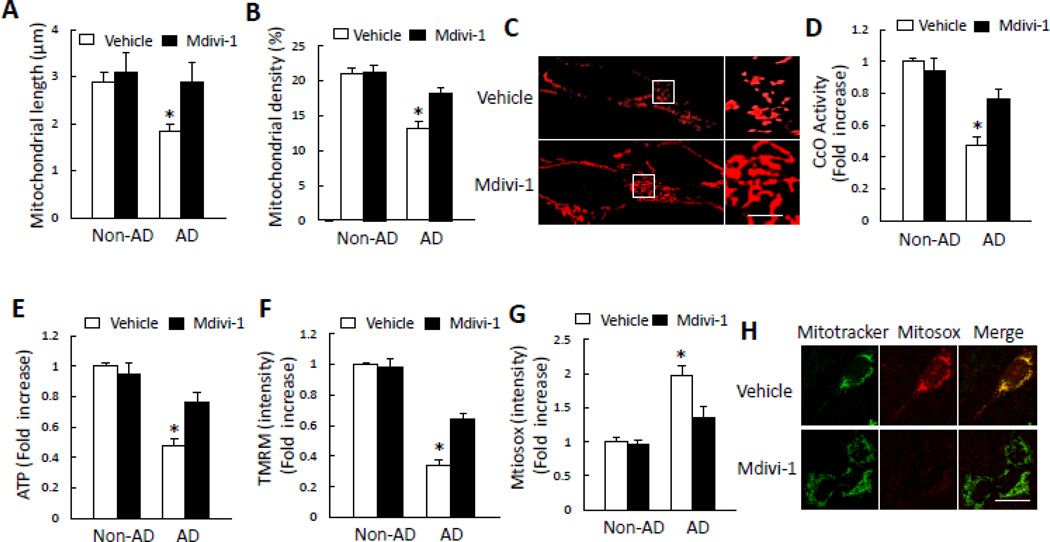
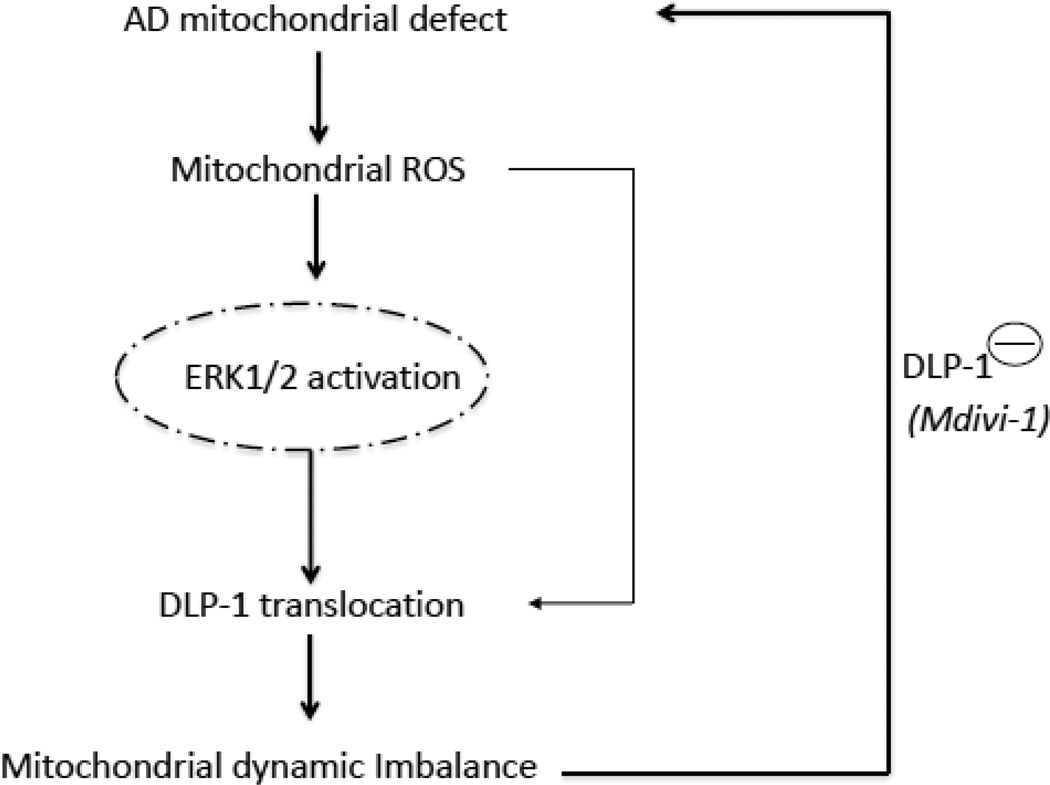
Mitochondrial dysfunction is an early pathological feature of Alzheimer's disease (AD). The underlying mechanisms and strategies to repair it remain unclear. Here, we demonstrate for the first time the direct consequences and potential mechanisms of mitochondrial functional defects associated with abnormal mitochondrial dynamics in AD. Using cytoplasmic hybrid (cybrid) neurons with incorporated platelet mitochondria from AD and age-matched non-AD human subjects into mitochondrial DNA (mtDNA)-depleted neuronal cells, we observed that AD cybrid cells had significant changes in morphology and function; such changes associate with altered expression and distribution of dynamin-like protein (DLP1) and mitofusin 2 (Mfn2). Treatment with antioxidant protects against AD mitochondria-induced extracellular signal-regulated kinase (ERK) activation and mitochondrial fission-fusion imbalances. Notably, inhibition of ERK activation not only attenuates aberrant mitochondrial morphology and function but also restores the mitochondrial fission and fusion balance. These effects suggest a role of oxidative stress-mediated ERK signal transduction in modulation of mitochondrial fission and fusion events. Further, blockade of the mitochondrial fission protein DLP1 by a genetic manipulation with a dominant negative DLP1 (DLP1(K38A)), its expression with siRNA-DLP1, or inhibition of mitochondrial division with mdivi-1 attenuates mitochondrial functional defects observed in AD cybrid cells. Our results provide new insights into mitochondrial dysfunction resulting from changes in the ERK-fission/fusion (DLP1) machinery and signaling pathway. The protective effect of mdivi-1 and inhibition of ERK signaling on maintenance of normal mitochondrial structure and function holds promise as a potential novel therapeutic strategy for AD.
Keywords: AD; Alzheimer's disease; Amyloid beta peptide; Aβ; CcO; Cybrid cells; Cytochrome c oxidase; DLP1; Dynamin-like protein; ERK; Extracellular signal-regulated kinase; HD; Huntington disease; Mfn2; Mitochondrial DNA; Mitochondrial fission and fusion; Mitofusin 2; PD; Parkinson disease; ROS; Reactive oxygen species; TMRM; Tetramethylrhodaminemethylester; mtDNA.
Copyright © 2013 Elsevier B.V. All rights reserved.
Conflict of interest statement
Figures



References
-
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
