

Rapidly progressive dementia with thalamic degeneration and peculiar cortical prion protein immunoreactivity, but absence of proteinase K resistant PrP: a new disease entity?
- PMID: 24252716
- PMCID: PMC3835463
- DOI: 10.1186/2051-5960-1-72
Rapidly progressive dementia with thalamic degeneration and peculiar cortical prion protein immunoreactivity, but absence of proteinase K resistant PrP: a new disease entity?
Abstract
Background: Human prion diseases are a group of rare fatal neurodegenerative conditions with well-developed clinical and neuropathological diagnostic criteria. Recent observations have expanded the spectrum of prion diseases beyond the classically recognized forms.
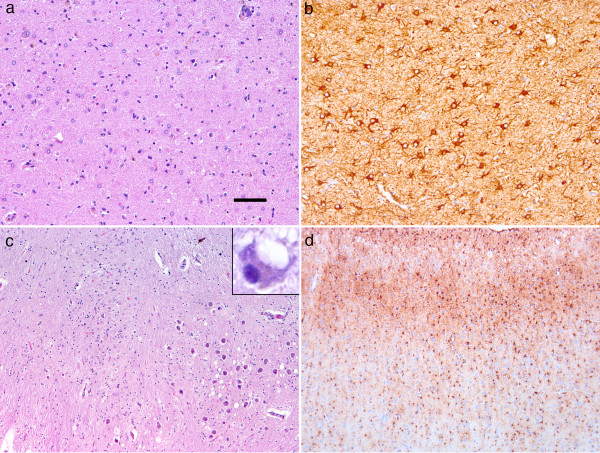
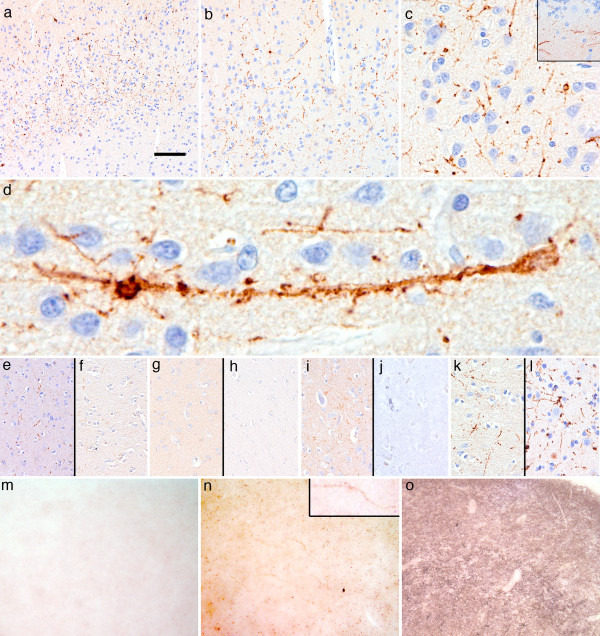
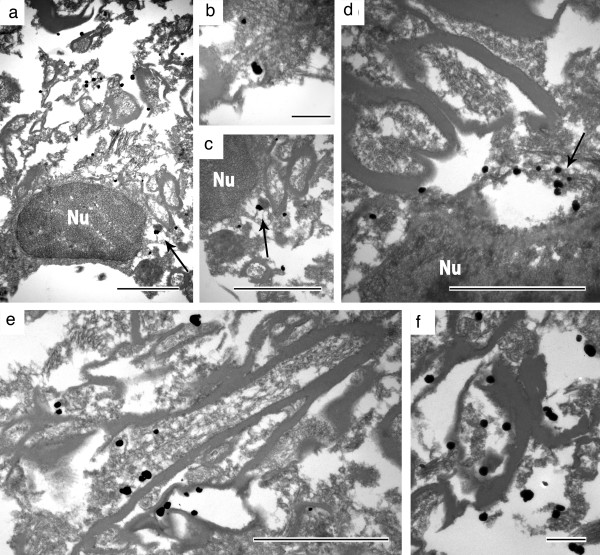
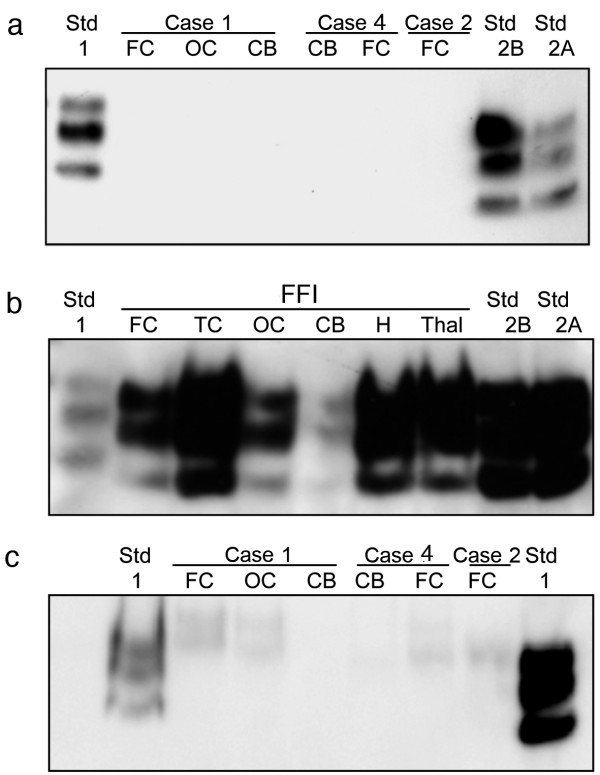
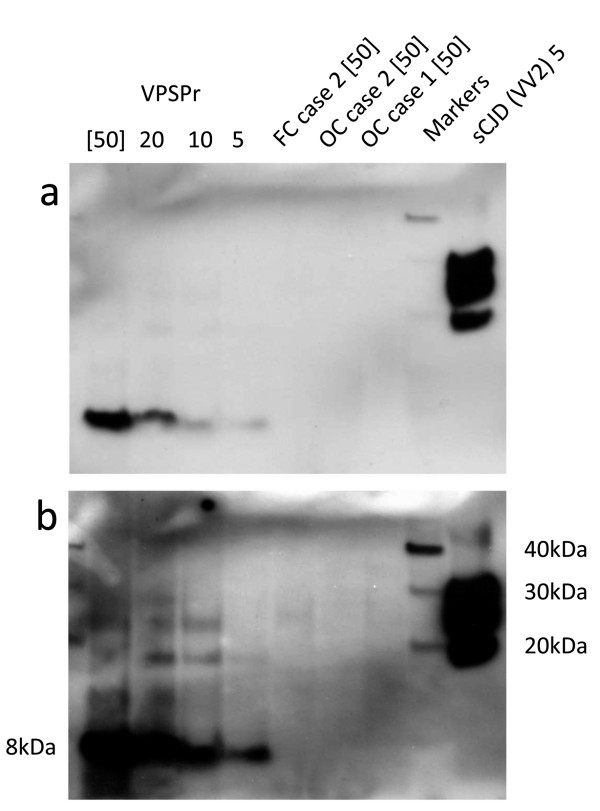
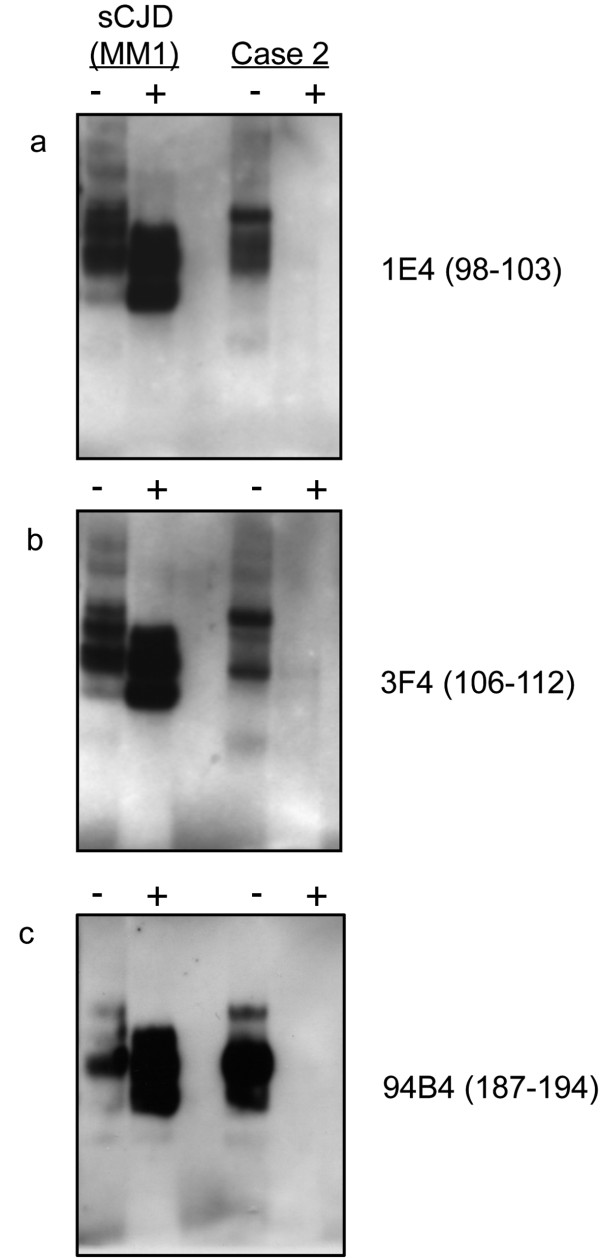
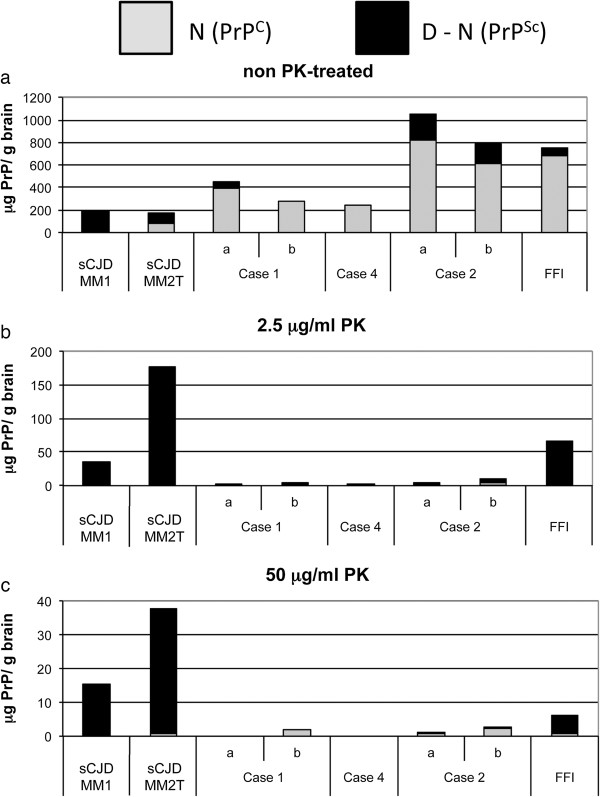
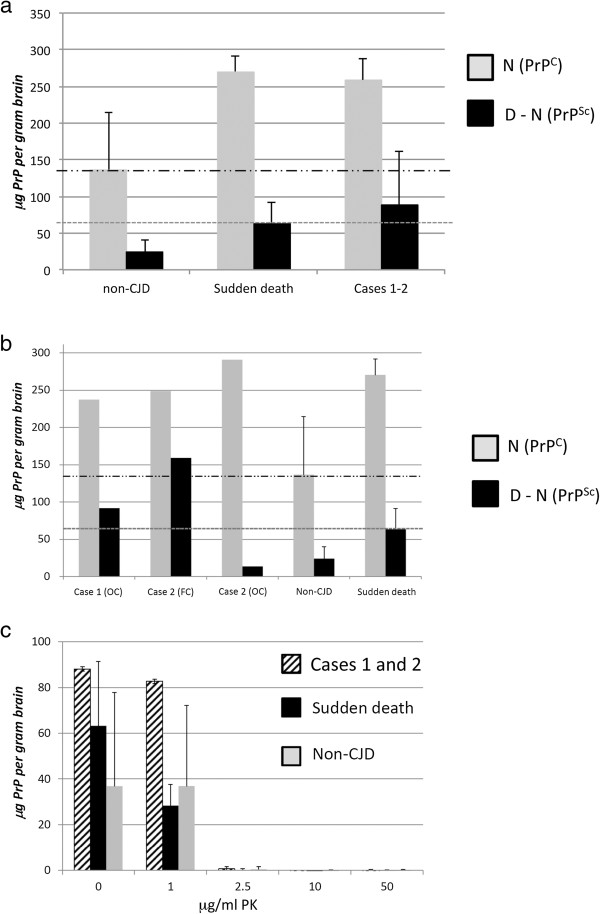
Results: In the present study we report six patients with a novel, apparently sporadic disease characterised by thalamic degeneration and rapidly progressive dementia (duration of illness 2-12 months; age at death: 55-81 years). Light and electron microscopic immunostaining for the prion protein (PrP) revealed a peculiar intraneuritic distribution in neocortical regions. Proteinase K resistant PrP (PrPres) was undetectable by Western blotting in frontal cortex from the three cases with frozen tissue, even after enrichment for PrPres by centrifugation or by phosphotungstic acid precipitation. Conformation-dependent immunoassay analysis using a range of PK digestion conditions (and no PK digestion) produced only very limited evidence of meaningful D-N (denatured/native) values, indicative of the presence of disease-associated PrP (PrPSc) in these cases, when the results were compared with appropriate negative control groups.
Conclusions: Our observation expands the spectrum of conditions associated with rapidly progressive dementia and may have implications for the understanding of the pathogenesis of prion diseases.
Figures
References
-
- Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IR. et al.Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57(10):979–988. doi: 10.1097/00005072-199810000-00010. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
