

In vivo modification of Abeta plaque toxicity as a novel neuroprotective lithium-mediated therapy for Alzheimer's disease pathology
- PMID: 24252759
- PMCID: PMC3833287
- DOI: 10.1186/2051-5960-1-73
In vivo modification of Abeta plaque toxicity as a novel neuroprotective lithium-mediated therapy for Alzheimer's disease pathology
Abstract
Background: Alzheimer's disease (AD) is characterized by the abnormal accumulation of extracellular beta-amyloid (Abeta) plaques, intracellular hyperphosphorylated tau, progressive synaptic alterations, axonal dystrophies, neuronal loss and the deterioration of cognitive capabilities of patients. However, no effective disease-modifying treatment has been yet developed. In this work we have evaluated whether chronic lithium treatment could ameliorate the neuropathology evolution of our well characterized PS1M146LxAPPSwe-London mice model.
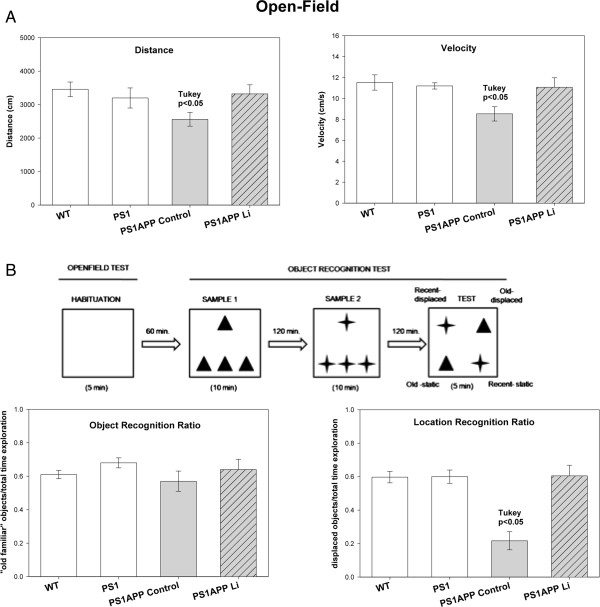
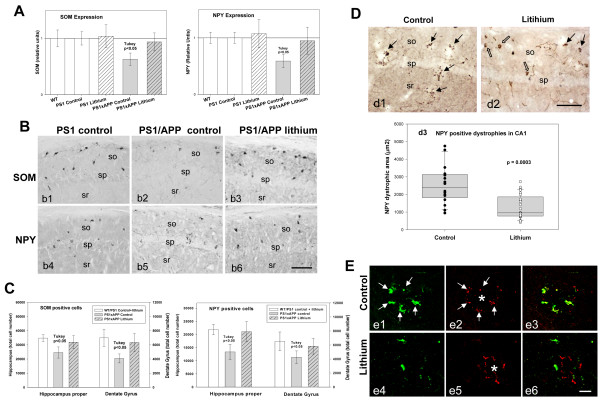
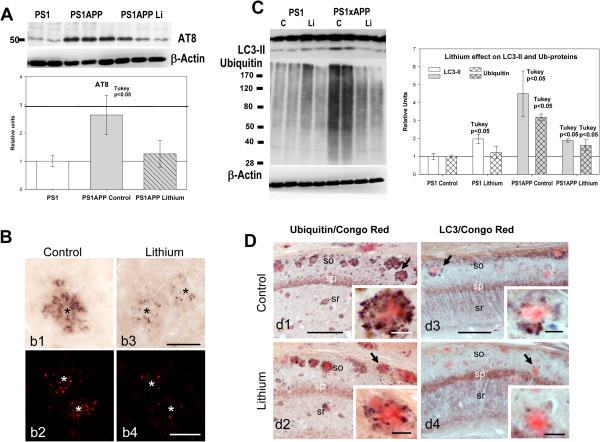
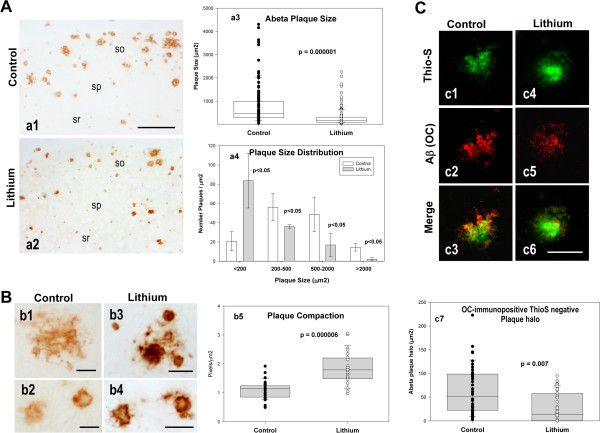
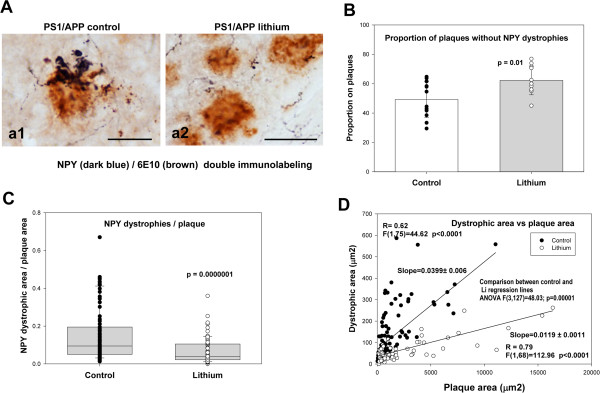
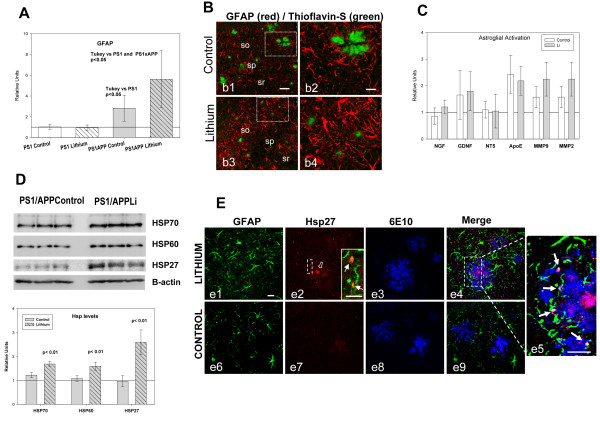
Results: Though beneficial effects of lithium have been previously described in different AD models, here we report a novel in vivo action of this compound that efficiently ameliorated AD-like pathology progression and rescued memory impairments by reducing the toxicity of Abeta plaques. Transgenic PS1M146LxAPPSwe-London mice, treated before the pathology onset, developed smaller plaques characterized by higher Abeta compaction, reduced oligomeric-positive halo and therefore with attenuated capacity to induce neuronal damage. Importantly, neuronal loss in hippocampus and entorhinal cortex was fully prevented. Our data also demonstrated that the axonal dystrophic area associated with lithium-modified plaques was highly reduced. Moreover, a significant lower accumulation of phospho-tau, LC3-II and ubiquitinated proteins was detected in treated mice. Our study highlights that this switch of plaque quality by lithium could be mediated by astrocyte activation and the release of heat shock proteins, which concentrate in the core of the plaques.
Conclusions: Our data demonstrate that the pharmacological in vivo modulation of the extracellular Abeta plaque compaction/toxicity is indeed possible and, in addition, might constitute a novel promising and innovative approach to develop a disease-modifying therapeutic intervention against AD.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
