

Molecular determinants of susceptibility to oncolytic vesicular stomatitis virus in pancreatic adenocarcinoma
- PMID: 24252853
- PMCID: PMC3959227
- DOI: 10.1016/j.jss.2013.10.032
Molecular determinants of susceptibility to oncolytic vesicular stomatitis virus in pancreatic adenocarcinoma
Abstract
Background: M protein mutant vesicular stomatitis virus (M51R-VSV) has oncolytic properties against many cancers. However, some cancer cells are resistant to M51R-VSV. Herein, we evaluate the molecular determinants of vesicular stomatitis virus (VSV) resistance in pancreatic adenocarcinoma cells.
Methods: Cell viability and the effect of β-interferon (IFN) were analyzed using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assay. Gene expression was evaluated via microarray analysis. Cell infectability was measured by flow cytometry. Xenografts were established in athymic nude mice and treated with intratumoral M51R-VSV.
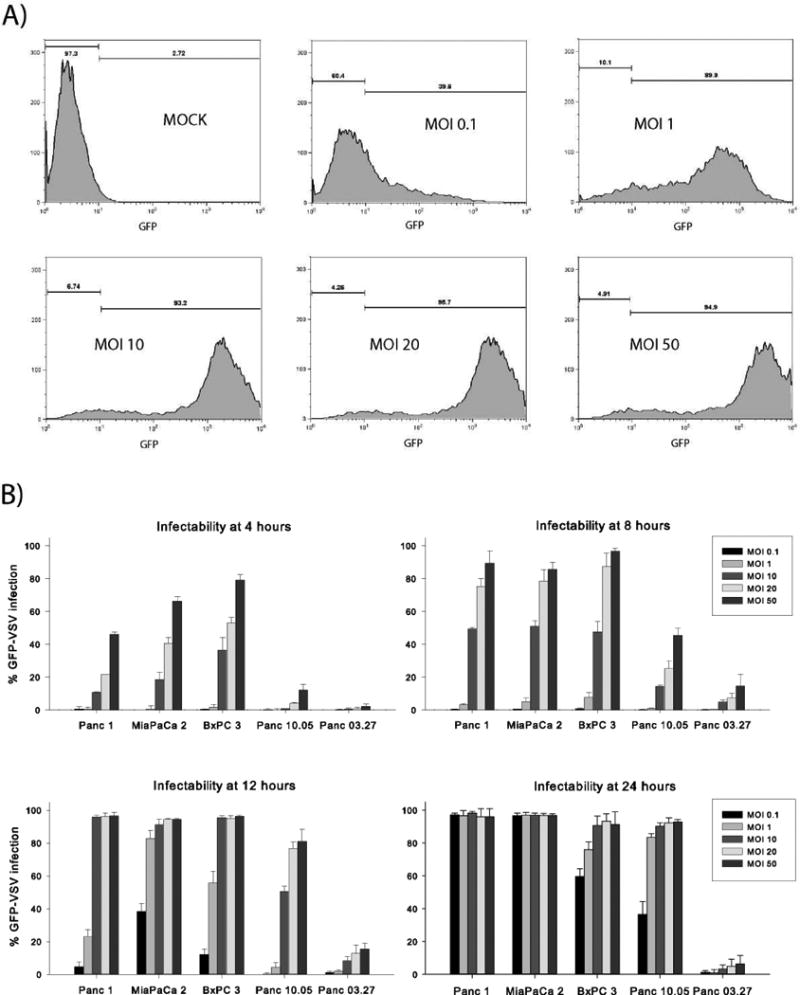
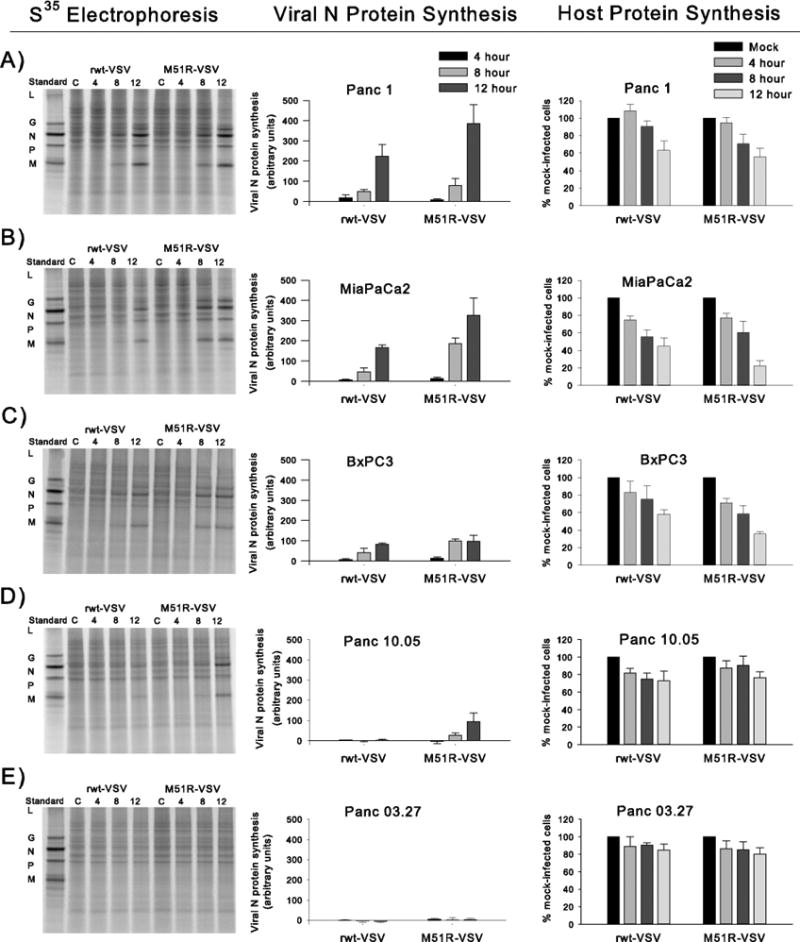
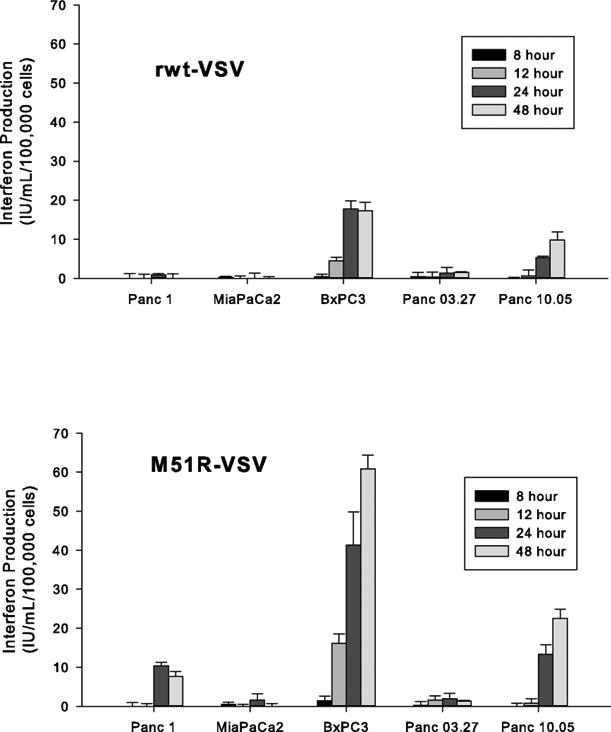
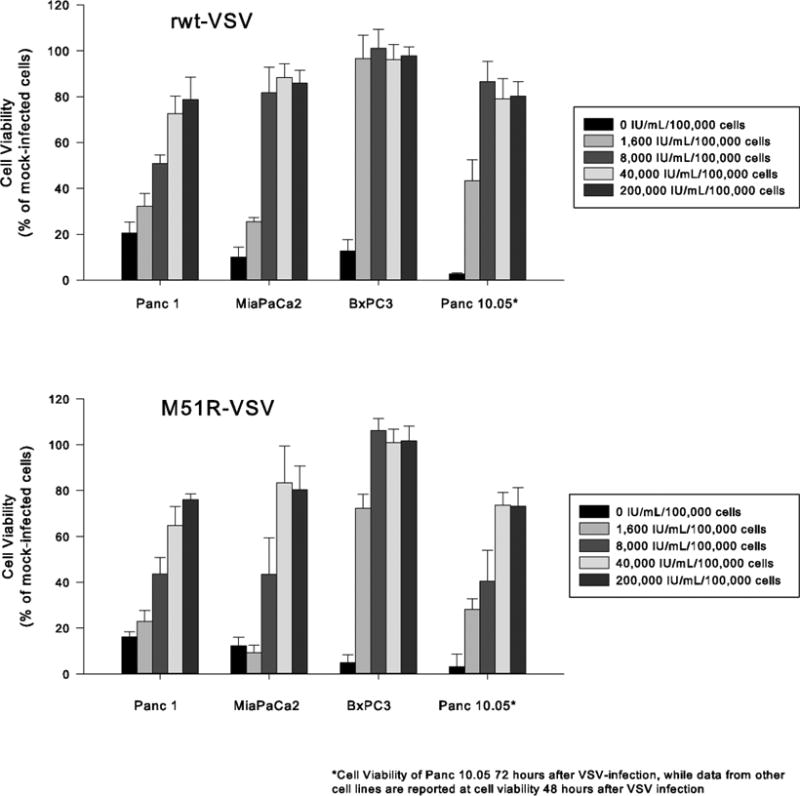
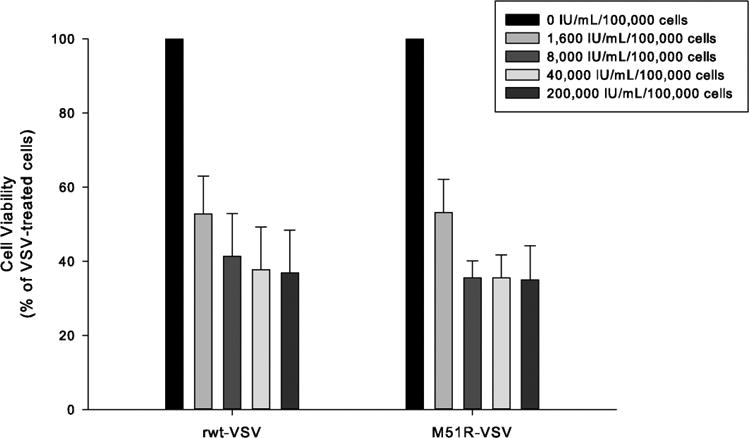
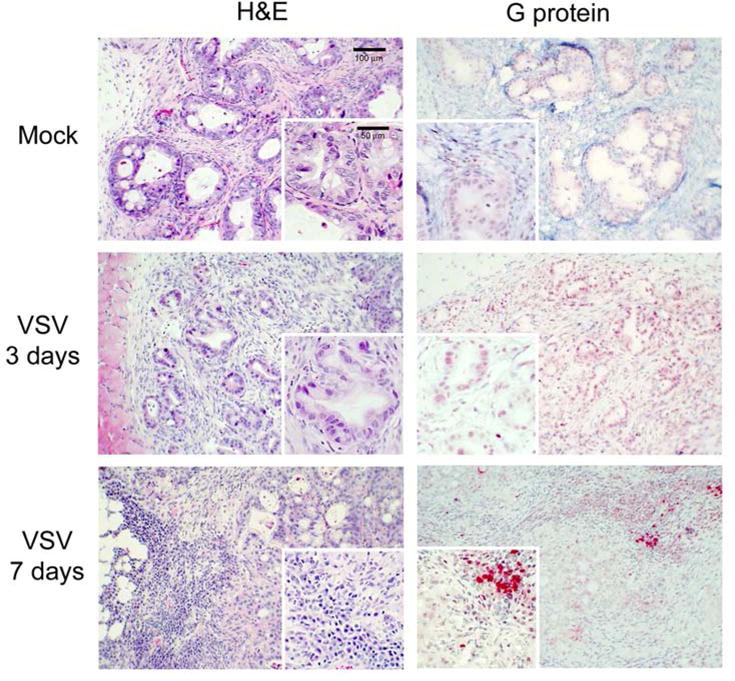
Results: Four of five pancreatic cancer cell lines were sensitive to M51R-VSV, whereas Panc 03.27 cells remained resistant (81 ± 3% viability 72 h after single-cycle infection). Comparing sensitive MiaPaCa2 cells with resistant Panc 03.27 cells, significant differences in gene expression were found relating to IFN signaling (P = 2 × 10(-5)), viral entry (P = 3 × 10(-4)), and endocytosis (P = 7 × 10(-4)). MiaPaCa2 cells permitted high levels of VSV infection, whereas Panc 03.27 cells were capable of resisting VSV cell entry even at high multiplicities of infection. Extrinsic β-IFN overcame apparent defects in IFN-mediated pathways in MiaPaCa2 cells conferring VSV resistance. In contrast, β-IFN decreased cell viability in Panc 3.27 cells, suggesting intact antiviral mechanisms. VSV-treated xenografts exhibited reduced tumor growth relative to controls in both MiaPaCa2 (1423 ± 345% versus 164 ± 136%; P < 0.001) and Panc 3.27 (979 ± 153% versus 50 ± 56%; P = 0.002) tumors. Significant lymphocytic infiltration was seen in M51R-VSV-treated Panc 03.27 xenografts.
Conclusions: Inhibition of VSV endocytosis and intact IFN-mediated defenses are responsible for M51R-VSV resistance in pancreatic adenocarcinoma cells. M51R-VSV treatment appears to induce antitumor cellular immunity in vivo, which may expand its clinical efficacy.
Keywords: Interferon; Pancreatic adenocarcinoma; Vesicular stomatitis virus; Viral endocytosis; Xenograft.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures




References
-
- Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 2004;17:516–27. - PubMed
-
- Ahmed M, Lyles DS. Identification of a consensus mutation in M protein of vesicular stomatitis virus from persistently infected cells that affects inhibition of host-directed gene expression. Virology. 1997;237:378–88. - PubMed
-
- Ebert O, Shinozaki K, Huang TG, et al. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Res. 2003;63:3605–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
