

p75 neurotrophin receptor and fenretinide-induced signaling in neuroblastoma
- PMID: 24253178
- PMCID: PMC3946654
- DOI: 10.1007/s00280-013-2355-y
p75 neurotrophin receptor and fenretinide-induced signaling in neuroblastoma
Abstract
Purpose: Neuroblastoma is the most common extracranial solid tumor of childhood. The retinoic acid analogue, fenretinide (4-hydroxyphenyl retinamide; 4-HPR), induces apoptosis in neuroblastoma cells in vitro and is currently in clinical trials for children with refractory neuroblastoma. We have previously shown that expression of the p75 neurotrophin receptor (p75NTR) enhances apoptosis induction and mitochondrial accumulation of reactive oxygen species by 4-HPR in neuroblastoma cells. We now examine the signaling events that underlie this effect.
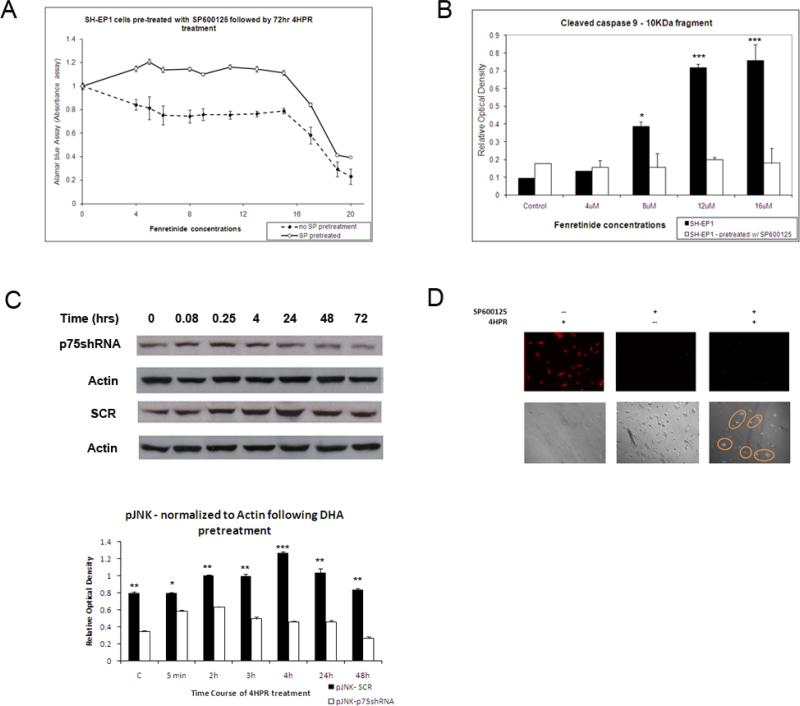
Methods: Systematic examination of pro- and anti-apoptotic signaling effectors was performed by Western blot. Specific inhibitors of JNK phosphorylation and scavengers of mitochondrial reactive oxygen species were used to demonstrate the roles of these phenomena in the enhancement of fenretinide efficacy.
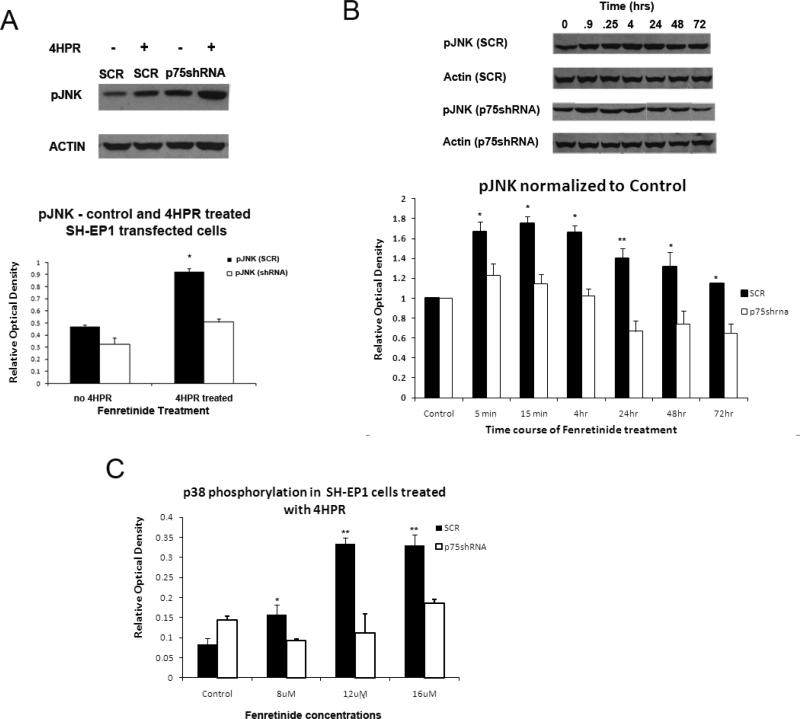
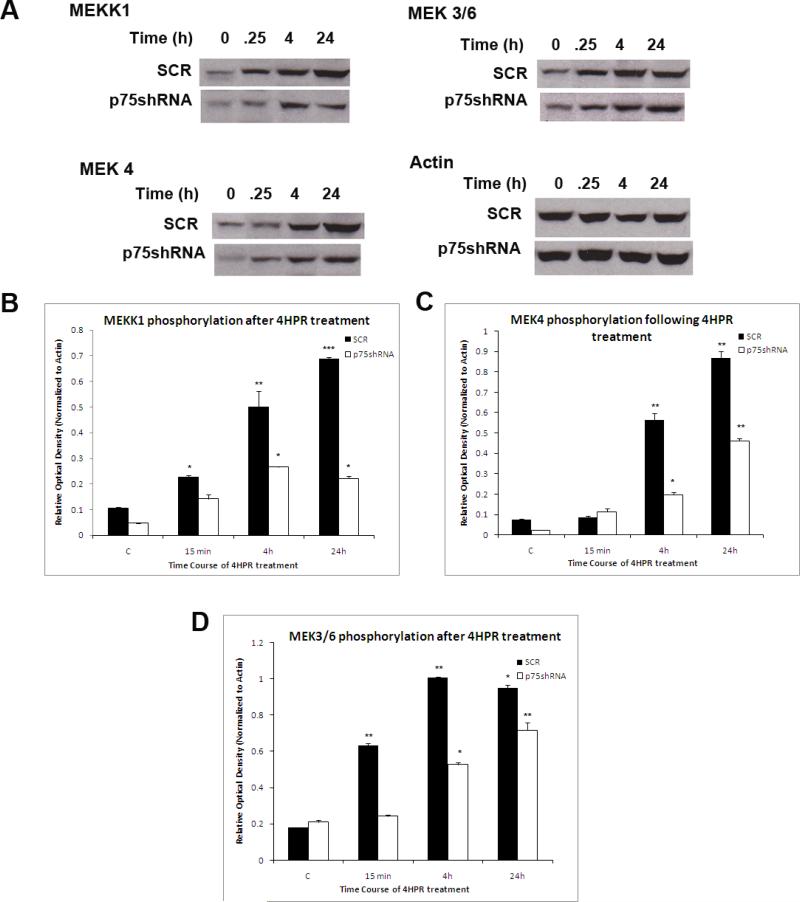
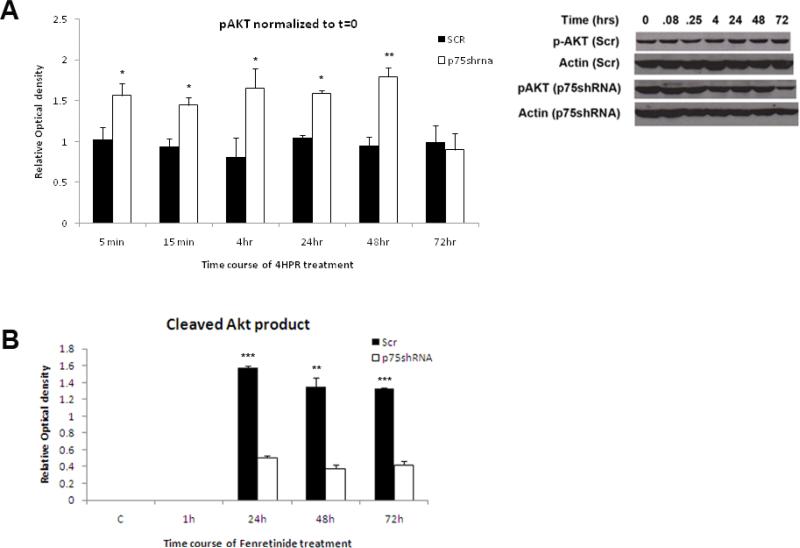
Results: The present studies demonstrate that enhancement of 4-HPR-induced apoptosis by p75NTR is dependent upon p38MAPK phosphorylation, JNK phosphorylation, caspase 3 activation, Akt cleavage, and decreased Akt phosphorylation. In addition, treatment with 4-HPR results in upregulation of MKK4 and MEKK1, and phosphorylation of MKK3/6. Efforts to enhance the efficacy of 4-HPR and to identify those tumors most likely to respond to it might exploit these effectors of 4-HPR-induced apoptosis.
Conclusions: Pharmacological agents that enhance MKK4 or MEKK1 expression or JNK expression or phosphorylation may enhance efficacy of 4-HPR in neuroblastomas that do not express high levels of p75NTR.
Figures
Similar articles
-
Cell Line-Dependent Variability of Coordinate Expression of p75NTR and CRABP1 and Modulation of Effects of Fenretinide on Neuroblastoma Cells.Oxid Med Cell Longev. 2016;2016:7568287. doi: 10.1155/2016/7568287. Epub 2015 Dec 30. Oxid Med Cell Longev. 2016. PMID: 26843908 Free PMC article.
-
p75NTR: an enhancer of fenretinide toxicity in neuroblastoma.Cancer Chemother Pharmacol. 2013 Mar;71(3):777-87. doi: 10.1007/s00280-013-2071-7. Epub 2013 Jan 13. Cancer Chemother Pharmacol. 2013. PMID: 23314735 Free PMC article.
-
4-oxo-fenretinide, a recently identified fenretinide metabolite, induces marked G2-M cell cycle arrest and apoptosis in fenretinide-sensitive and fenretinide-resistant cell lines.Cancer Res. 2006 Mar 15;66(6):3238-47. doi: 10.1158/0008-5472.CAN-05-3362. Cancer Res. 2006. PMID: 16540676
-
Molecular mechanisms of fenretinide-induced apoptosis of neuroblastoma cells.Ann N Y Acad Sci. 2004 Dec;1028:81-9. doi: 10.1196/annals.1322.009. Ann N Y Acad Sci. 2004. PMID: 15650234 Review.
-
Induction of GADD153 and Bak: novel molecular targets of fenretinide-induced apoptosis of neuroblastoma.Cancer Lett. 2003 Jul 18;197(1-2):157-63. doi: 10.1016/s0304-3835(03)00098-3. Cancer Lett. 2003. PMID: 12880976 Review.
Cited by
-
Stem Cell Markers in Neuroblastoma-An Emerging Role for LGR5.Front Cell Dev Biol. 2015 Dec 2;3:77. doi: 10.3389/fcell.2015.00077. eCollection 2015. Front Cell Dev Biol. 2015. PMID: 26697427 Free PMC article.
-
Induction of Expression of p75 Neurotrophin Receptor Intracellular Domain Does Not Induce Expression or Enhance Activity of Mitochondrial Complex II.Oxid Med Cell Longev. 2016;2016:8752821. doi: 10.1155/2016/8752821. Epub 2015 Nov 10. Oxid Med Cell Longev. 2016. PMID: 26640617 Free PMC article.
-
Cell Line-Dependent Variability of Coordinate Expression of p75NTR and CRABP1 and Modulation of Effects of Fenretinide on Neuroblastoma Cells.Oxid Med Cell Longev. 2016;2016:7568287. doi: 10.1155/2016/7568287. Epub 2015 Dec 30. Oxid Med Cell Longev. 2016. PMID: 26843908 Free PMC article.
References
-
- Lovat PE, Corazzari M, Goranov B, et al. Molecular mechanisms of fenretinide-induced apoptosis of neuroblastoma cells. Ann N Y Acad Sci. 2004;1028:81–89. - PubMed
-
- Lovat PE, Ranalli M, Corazzari M, et al. Mechanisms of free-radical induction in relation to fenretinide-induced apoptosis of neuroblastoma. J Cell Biochem. 2003;89:698–708. - PubMed
-
- Lovat PE, Corazzari M, Di Sano F, et al. The role of gangliosides in fenretinide-induced apoptosis of neuroblastoma. Cancer Lett. 2005;228:105–110. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
