

Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2
- PMID: 24253200
- PMCID: PMC3891447
- DOI: 10.1093/brain/awt315
Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2
Abstract
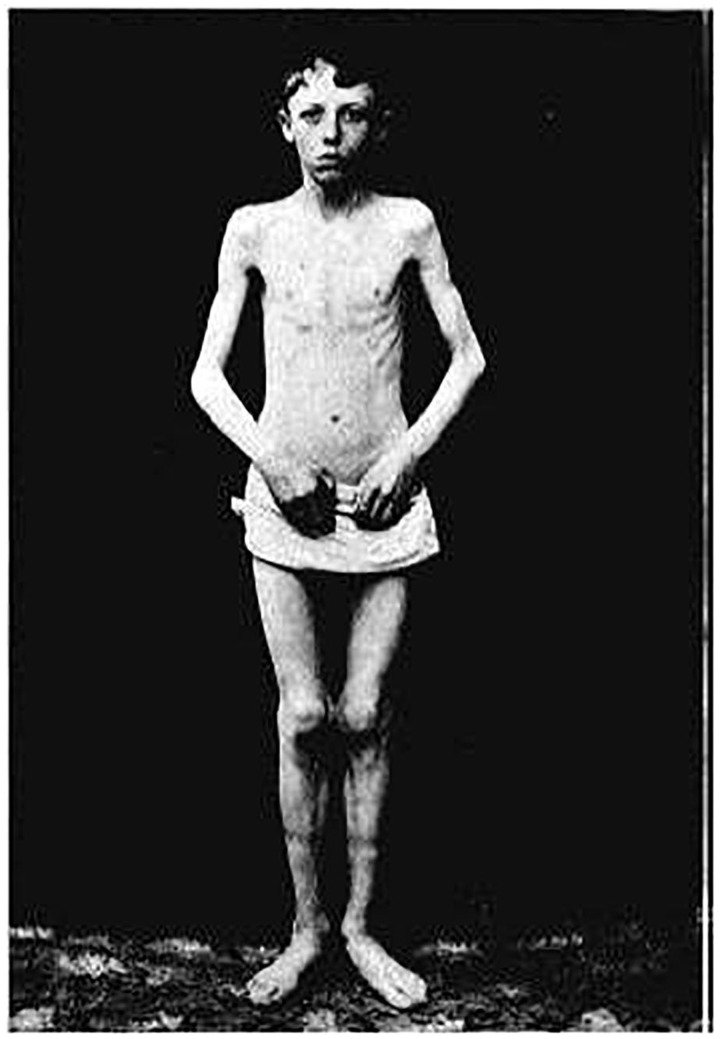
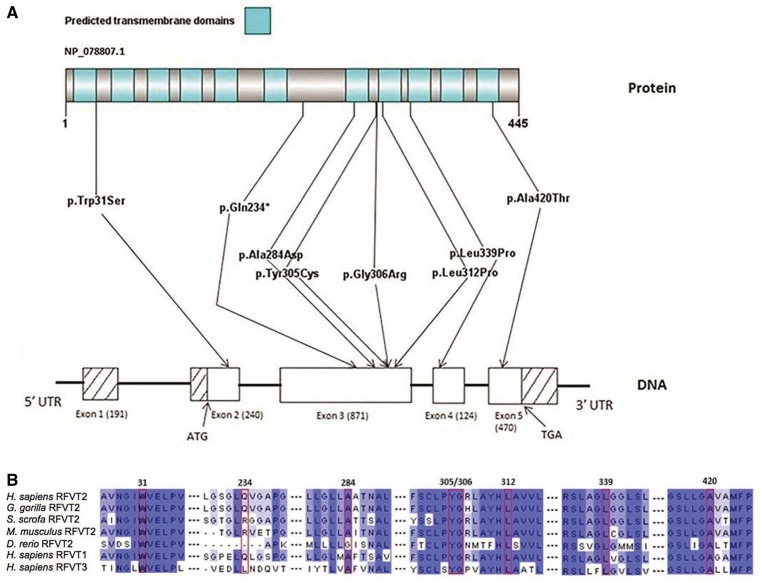
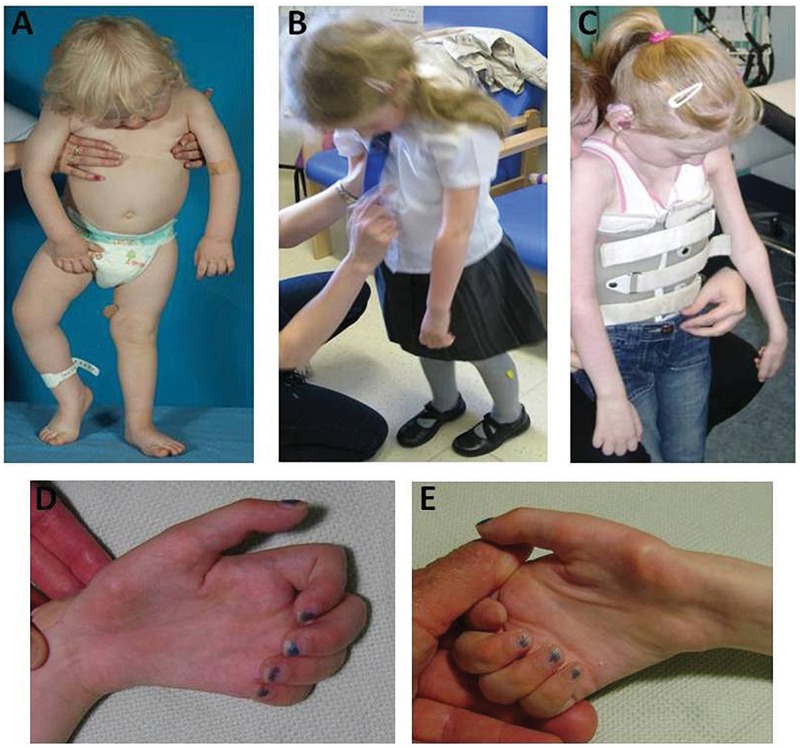
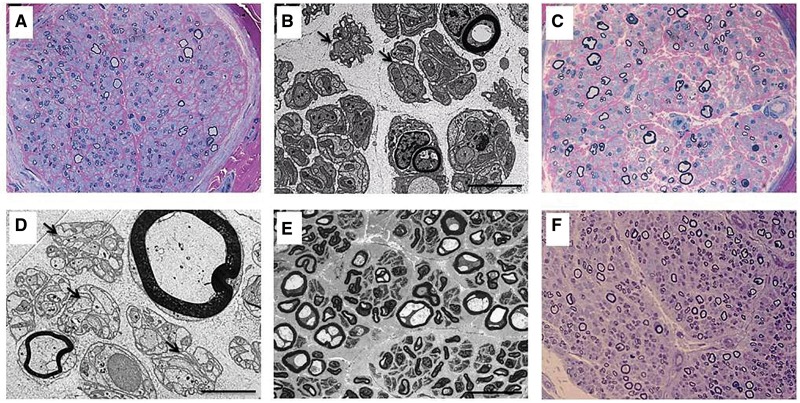
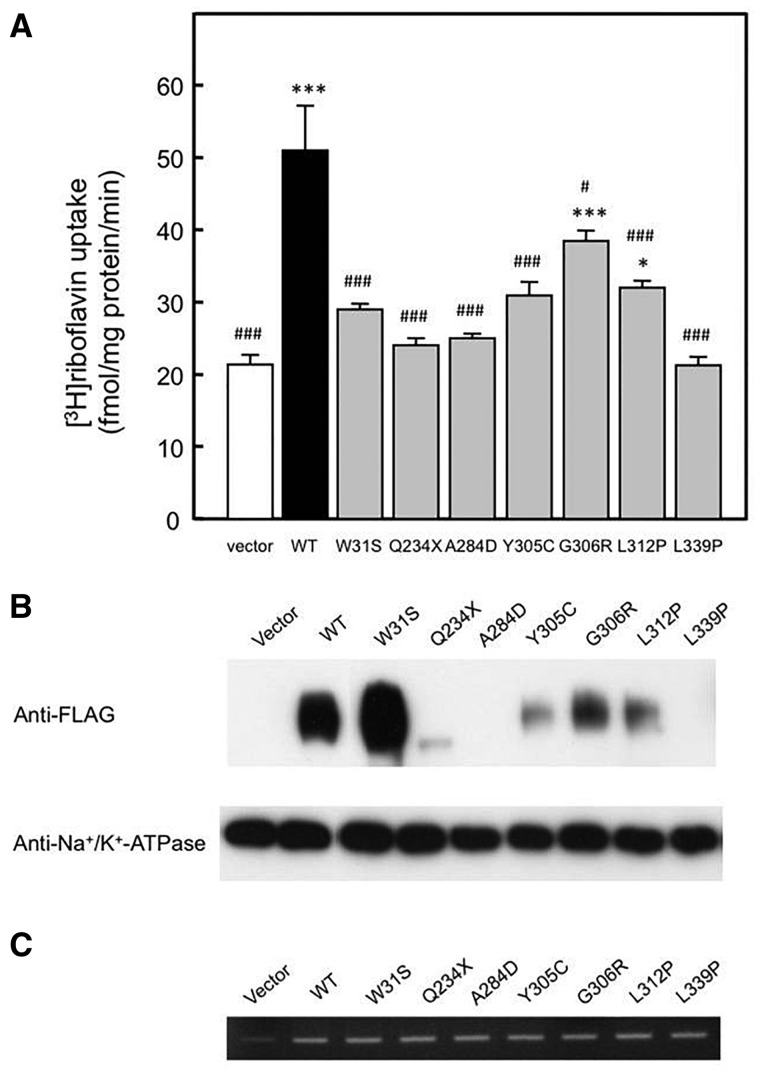
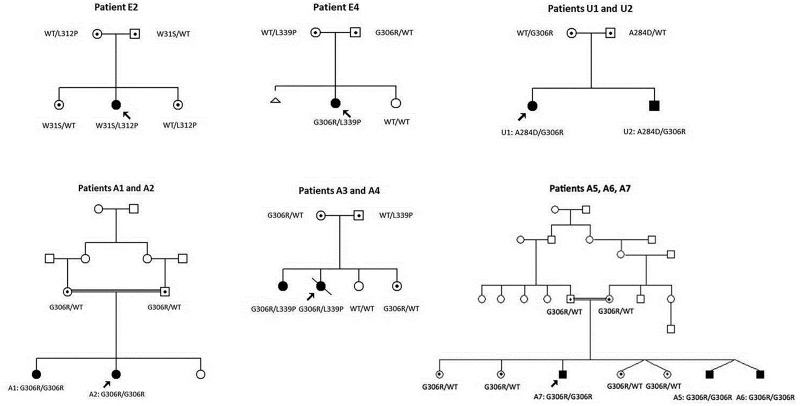
Childhood onset motor neuron diseases or neuronopathies are a clinically heterogeneous group of disorders. A particularly severe subgroup first described in 1894, and subsequently called Brown-Vialetto-Van Laere syndrome, is characterized by progressive pontobulbar palsy, sensorineural hearing loss and respiratory insufficiency. There has been no treatment for this progressive neurodegenerative disorder, which leads to respiratory failure and usually death during childhood. We recently reported the identification of SLC52A2, encoding riboflavin transporter RFVT2, as a new causative gene for Brown-Vialetto-Van Laere syndrome. We used both exome and Sanger sequencing to identify SLC52A2 mutations in patients presenting with cranial neuropathies and sensorimotor neuropathy with or without respiratory insufficiency. We undertook clinical, neurophysiological and biochemical characterization of patients with mutations in SLC52A2, functionally analysed the most prevalent mutations and initiated a regimen of high-dose oral riboflavin. We identified 18 patients from 13 families with compound heterozygous or homozygous mutations in SLC52A2. Affected individuals share a core phenotype of rapidly progressive axonal sensorimotor neuropathy (manifesting with sensory ataxia, severe weakness of the upper limbs and axial muscles with distinctly preserved strength of the lower limbs), hearing loss, optic atrophy and respiratory insufficiency. We demonstrate that SLC52A2 mutations cause reduced riboflavin uptake and reduced riboflavin transporter protein expression, and we report the response to high-dose oral riboflavin therapy in patients with SLC52A2 mutations, including significant and sustained clinical and biochemical improvements in two patients and preliminary clinical response data in 13 patients with associated biochemical improvements in 10 patients. The clinical and biochemical responses of this SLC52A2-specific cohort suggest that riboflavin supplementation can ameliorate the progression of this neurodegenerative condition, particularly when initiated soon after the onset of symptoms.
Keywords: Brown-Vialetto-Van Laere syndrome; RFVT2; SLC52A2; childhood neuronopathy; riboflavin therapy.
Figures
Comment in
-
Promising riboflavin treatment for motor neuron disorder.Brain. 2014 Jan;137(Pt 1):2-3. doi: 10.1093/brain/awt342. Brain. 2014. PMID: 24424912 No abstract available.
References
-
- Alhadeff L, Gualtieri CT, Lipton M. Toxic effects of water-soluble vitamins. Nutr Rev. 1984;42:33–40. - PubMed
-
- Anand G, Hasan N, Jayapal S, Huma Z, Ali T, Hull J, et al. Early use of high-dose riboflavin in a case of Brown-Vialetto-Van Laere syndrome. Dev Med Child Neurol. 2012;54:187–9. - PubMed
-
- Bosch AM, Abeling NG, Ijlst L, Knoester H, van der Pol WL, Stroomer AE, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34:159–64. - PMC - PubMed
-
- Brown C. Infantile amyotrophic lateral sclerosis of the family type. J Nerv Ment Dis. 1894;19:707–16.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- G0802760/MRC_/Medical Research Council/United Kingdom
- G1001253/MRC_/Medical Research Council/United Kingdom
- R01NS072248/NS/NINDS NIH HHS/United States
- 096919/WT_/Wellcome Trust/United Kingdom
- U54 NS065712/NS/NINDS NIH HHS/United States
- MR/K000608/1/MRC_/Medical Research Council/United Kingdom
- G0601943/MRC_/Medical Research Council/United Kingdom
- R01NS075764-01A1/NS/NINDS NIH HHS/United States
- G108/638/MRC_/Medical Research Council/United Kingdom
- MR/J004758/1/MRC_/Medical Research Council/United Kingdom
- U54NS065712/NS/NINDS NIH HHS/United States
- R01 NS072248/NS/NINDS NIH HHS/United States
- R01 NS075764/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
