

Continent-wide panmixia of an African fruit bat facilitates transmission of potentially zoonotic viruses
- PMID: 24253424
- PMCID: PMC3836177
- DOI: 10.1038/ncomms3770
Continent-wide panmixia of an African fruit bat facilitates transmission of potentially zoonotic viruses
Abstract
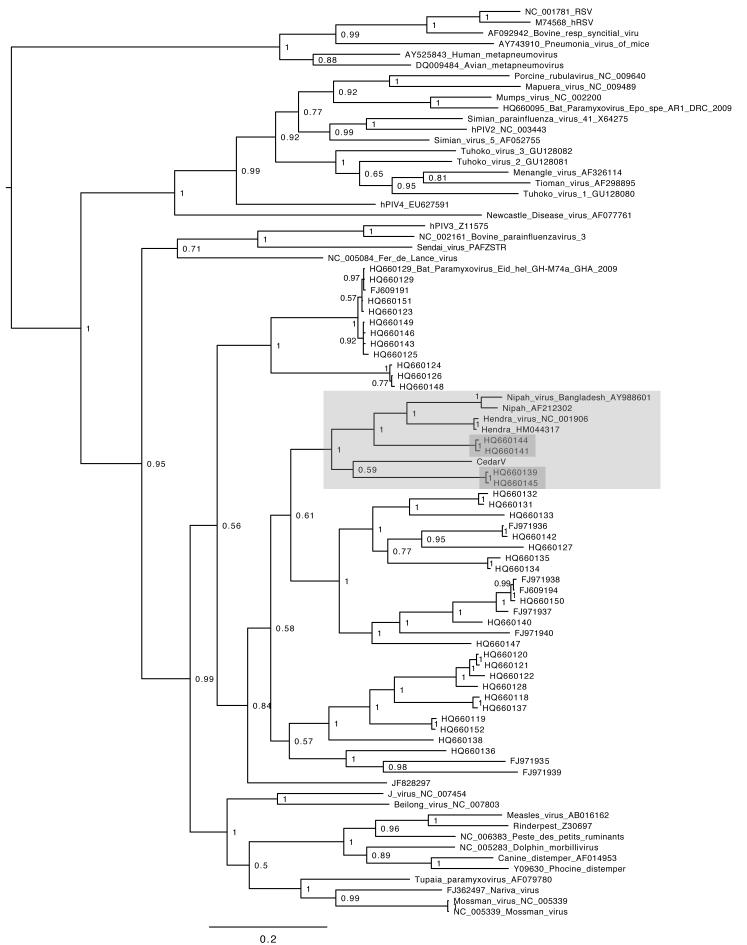
The straw-coloured fruit bat, Eidolon helvum, is Africa's most widely distributed and commonly hunted fruit bat, often living in close proximity to human populations. This species has been identified as a reservoir of potentially zoonotic viruses, but uncertainties remain regarding viral transmission dynamics and mechanisms of persistence. Here we combine genetic and serological analyses of populations across Africa, to determine the extent of epidemiological connectivity among E. helvum populations. Multiple markers reveal panmixia across the continental range, at a greater geographical scale than previously recorded for any other mammal, whereas populations on remote islands were genetically distinct. Multiple serological assays reveal antibodies to henipaviruses and Lagos bat virus in all locations, including small isolated island populations, indicating that factors other than population size and connectivity may be responsible for viral persistence. Our findings have potentially important public health implications, and highlight a need to avoid disturbances that may precipitate viral spillover.
Figures





Comment in
-
Bat trait, genetic and pathogen data from large-scale investigations of African fruit bats, Eidolon helvum.Sci Data. 2016 Aug 1;3:160049. doi: 10.1038/sdata.2016.49. Sci Data. 2016. PMID: 27479120 Free PMC article.
References
-
- Banyard AC, Hayman DTS, Johnson N, McElhinney LM, Fooks AR. Bats and lyssaviruses. Adv. Virus Res. 2011;79:239–289. - PubMed
-
- Mickleburgh S, Hutson A, Bergmans W, Fahr J, Racey PA. Eidolon helvum. In: IUCN, editor. 2008 IUCN Red List of Threatened Species. 2008. Available from: www.iucnredlist.org. Downloaded on 6 February 2011.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
