

Tumour suppressor gene function in carcinoma-associated fibroblasts: from tumour cells via EMT and back again?
- PMID: 24254977
- PMCID: PMC6664431
- DOI: 10.1002/path.4298
Tumour suppressor gene function in carcinoma-associated fibroblasts: from tumour cells via EMT and back again?
Abstract
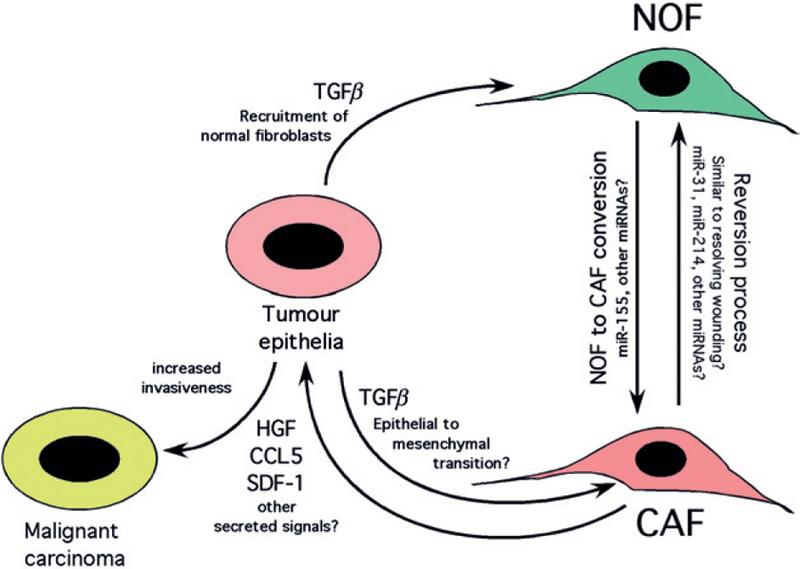
Recent reports indicate that inactivation of the RB, TP53 or PTEN tumour suppressor genes is detected in tumour stroma of oropharyngeal, breast and other human cancers. Mouse models have validated the tumour-promoting effects of deleting Rb, Pten or p53 in fibroblasts that converts them from normal fibroblasts to carcinoma associated fibroblasts (CAFs). The tumour-promoting activity of CAFs in these contexts was associated with increased paracrine signaling to tumour cells through production of specific growth factors, chemokines and MMPs by CAFs. The conversion of NOFs into CAFs through acquisition of specific mutations, such as loss of tumour suppressors, or deregulated expression of microRNAs or key epigenetic events, can clearly occur independently of genetic and epigenetic changes in tumour cells but an alternative source of CAFs that is being reconsidered is that CAFs derive from the tumour cells by EMT. Recent mouse models employing lineage-tracing techniques have suggested that this can take place in vivo and the extent to which this is relevant more broadly is discussed.
Keywords: EMT; Pten; RB; TGFβ; carcinoma-associated fibroblasts; lineage tracing; micro-RNAs; p53; trophic support.
Copyright © 2013 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Conflict of interest statement
No conflicts of interest were declared.
Figures

References
-
- Coussens LM, Hanahan D. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012; 21: 309–322. - PubMed
-
- Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer 2008; 8: 56–61. - PubMed
-
- Moinfar F, Man YG, Arnould L, et al. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res 2000; 60: 2562–2566. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
