

Aberrantly spliced HTT, a new player in Huntington's disease pathogenesis
- PMID: 24256709
- PMCID: PMC3907474
- DOI: 10.4161/rna.26706
Aberrantly spliced HTT, a new player in Huntington's disease pathogenesis
Abstract
Huntington's disease (HD) is an adult-onset neurodegenerative disorder caused by a mutated CAG repeat in the huntingtin gene that is translated into an expanded polyglutamine tract. The clinical manifestation of HD is a progressive physical, cognitive, and psychiatric deterioration that is eventually fatal. The mutant huntingtin protein is processed into several smaller fragments, which have been implicated as critical factors in HD pathogenesis. The search for proteases responsible for their production has led to the identification of several cleavage sites on the huntingtin protein. However, the origin of the small N-terminal fragments that are found in HD postmortem brains has remained elusive. Recent mapping of huntingtin fragments in a mouse model demonstrated that the smallest N-terminal fragment is an exon 1 protein. This discovery spurred our hypothesis that mis-splicing as opposed to proteolysis could be generating the smallest huntingtin fragment. We demonstrated that mis-splicing of mutant huntingtin intron 1 does indeed occur and results in a short polyadenylated mRNA, which is translated into an exon 1 protein. The exon 1 protein fragment is highly pathogenic. Transgenic mouse models containing just human huntingtin exon 1 develop a rapid onset of HD-like symptoms. Our finding that a small, mis-spliced HTT transcript and corresponding exon 1 protein are produced in the context of an expanded CAG repeat has unraveled a new molecular mechanism in HD pathogenesis. Here we present detailed models of how mis-splicing could be facilitated, what challenges remain in this model, and implications for therapeutic studies.
Keywords: HTT exon 1; Huntington’s disease; SRSF6; huntingtin fragment; mis-splicing.
Figures
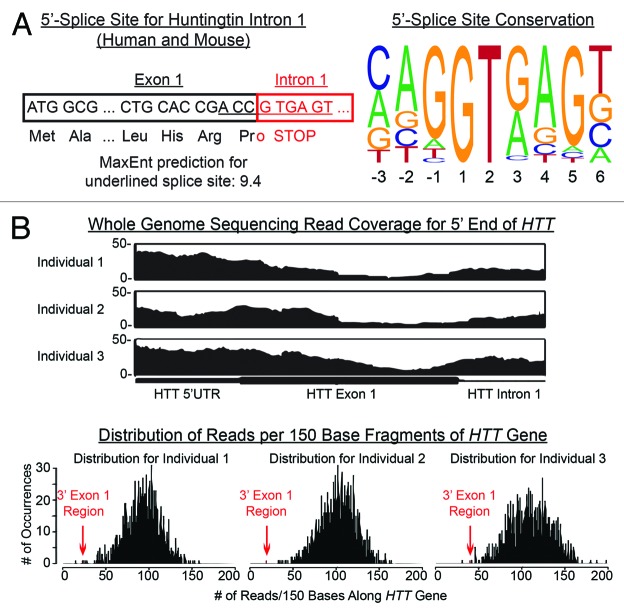
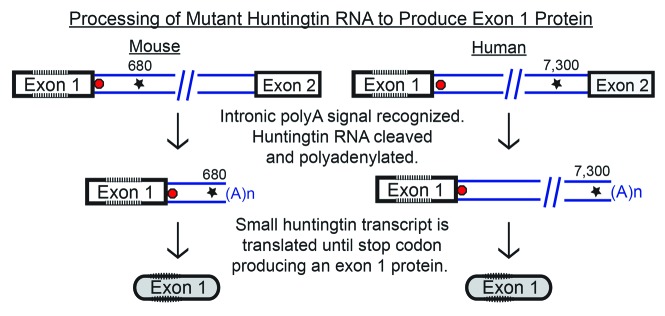
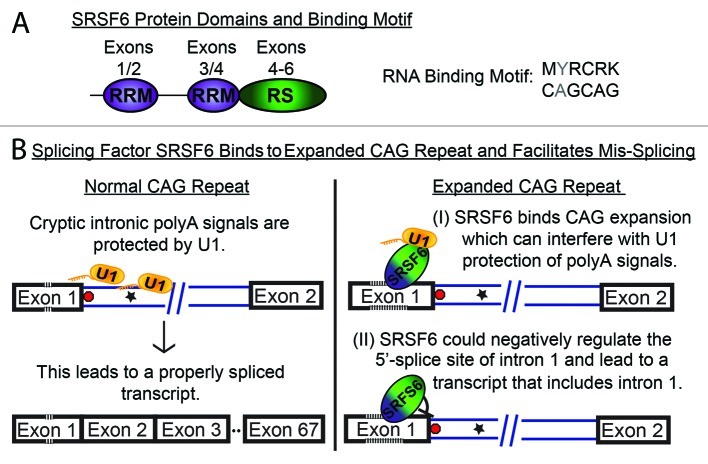
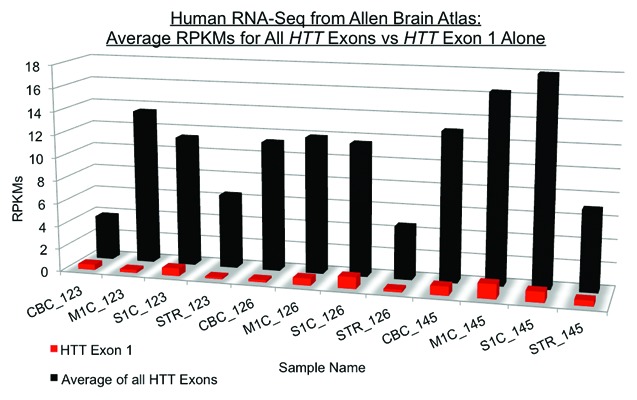
Comment on
-
Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease.Proc Natl Acad Sci U S A. 2013 Feb 5;110(6):2366-70. doi: 10.1073/pnas.1221891110. Epub 2013 Jan 22. Proc Natl Acad Sci U S A. 2013. PMID: 23341618 Free PMC article.
References
-
- Bates G, Harper PS, Jones L. Huntington's Disease Oxford University Press; 2002.
-
- Lunkes A, Lindenberg KS, Ben-Haïem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–69. doi: 10.1016/S1097-2765(02)00602-0. - DOI - PubMed
-
- Landles C, Sathasivam K, Weiss A, Woodman B, Moffitt H, Finkbeiner S, Sun B, Gafni J, Ellerby LM, Trottier Y, et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J Biol Chem. 2010;285:8808–23. doi: 10.1074/jbc.M109.075028. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical