

Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis
- PMID: 24256728
- PMCID: PMC4142699
- DOI: 10.1038/nature12681
Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis
Abstract
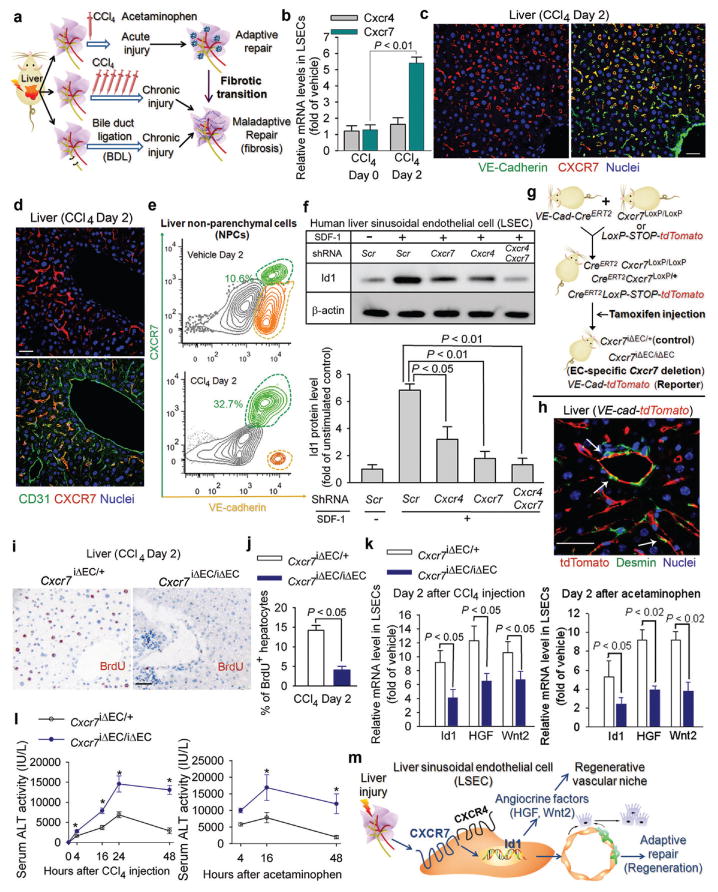
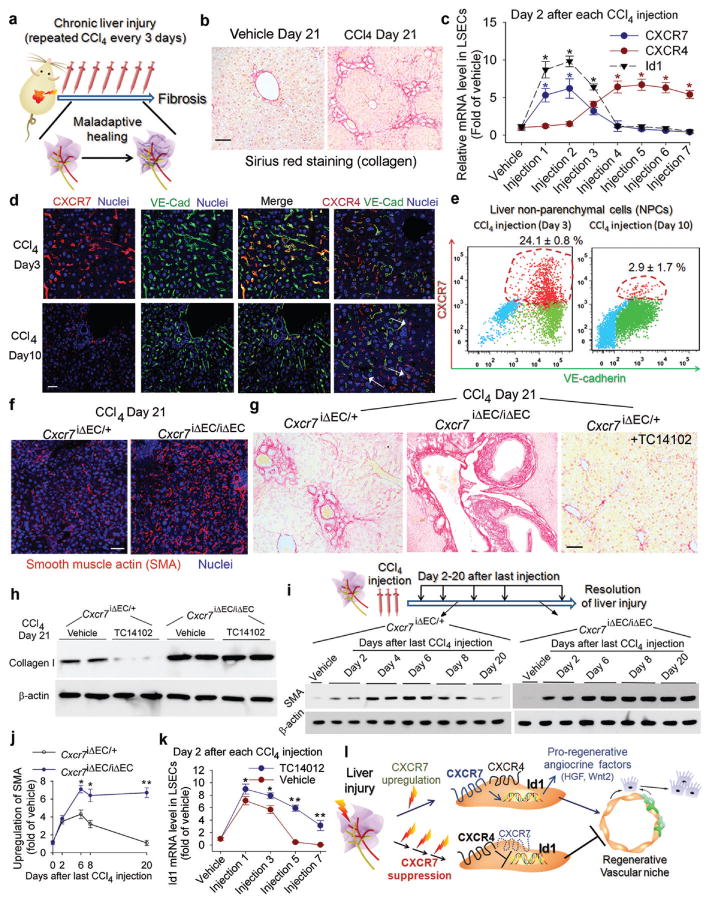
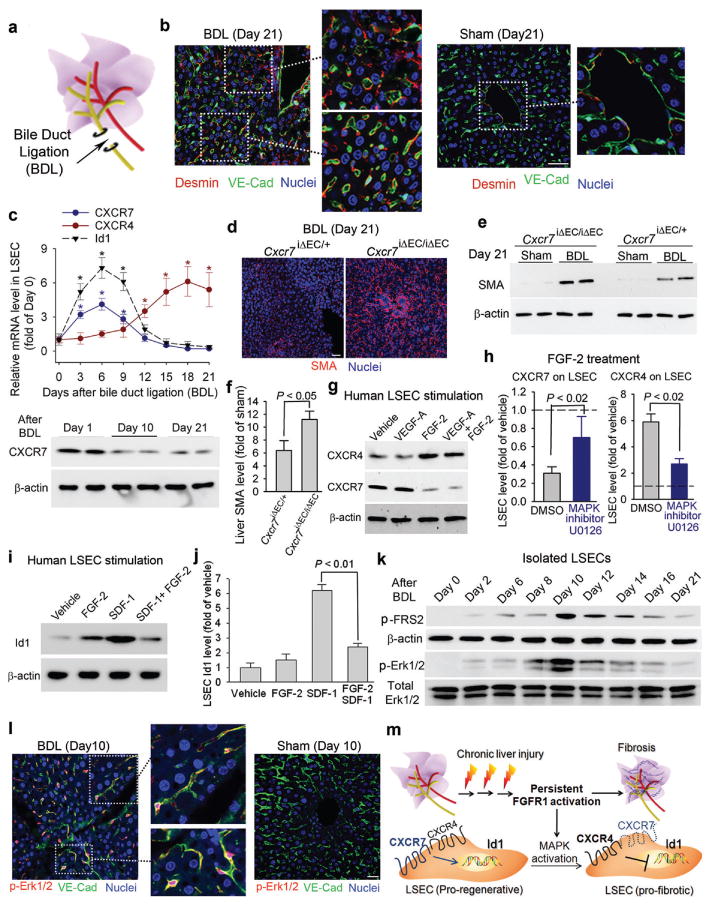
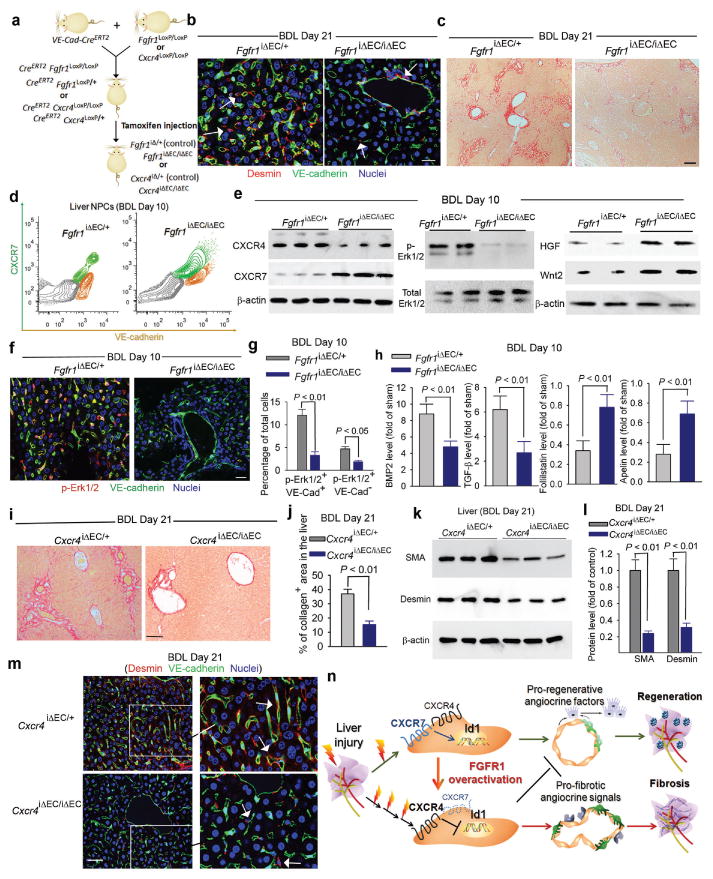
Chemical or traumatic damage to the liver is frequently associated with aberrant healing (fibrosis) that overrides liver regeneration. The mechanism by which hepatic niche cells differentially modulate regeneration and fibrosis during liver repair remains to be defined. Hepatic vascular niche predominantly represented by liver sinusoidal endothelial cells deploys paracrine trophogens, known as angiocrine factors, to stimulate regeneration. Nevertheless, it is not known how pro-regenerative angiocrine signals from liver sinusoidal endothelial cells is subverted to promote fibrosis. Here, by combining an inducible endothelial-cell-specific mouse gene deletion strategy and complementary models of acute and chronic liver injury, we show that divergent angiocrine signals from liver sinusoidal endothelial cells stimulate regeneration after immediate injury and provoke fibrosis after chronic insult. The pro-fibrotic transition of vascular niche results from differential expression of stromal-derived factor-1 receptors, CXCR7 and CXCR4 (refs 18, 19, 20, 21), in liver sinusoidal endothelial cells. After acute injury, CXCR7 upregulation in liver sinusoidal endothelial cells acts with CXCR4 to induce transcription factor Id1, deploying pro-regenerative angiocrine factors and triggering regeneration. Inducible deletion of Cxcr7 in sinusoidal endothelial cells (Cxcr7(iΔEC/iΔEC)) from the adult mouse liver impaired liver regeneration by diminishing Id1-mediated production of angiocrine factors. By contrast, after chronic injury inflicted by iterative hepatotoxin (carbon tetrachloride) injection and bile duct ligation, constitutive FGFR1 signalling in liver sinusoidal endothelial cells counterbalanced CXCR7-dependent pro-regenerative response and augmented CXCR4 expression. This predominance of CXCR4 over CXCR7 expression shifted angiocrine response of liver sinusoidal endothelial cells, stimulating proliferation of desmin(+) hepatic stellate-like cells and enforcing a pro-fibrotic vascular niche. Endothelial-cell-specific ablation of either Fgfr1 (Fgfr1(iΔEC/iΔEC)) or Cxcr4 (Cxcr4(iΔEC/iΔEC)) in mice restored the pro-regenerative pathway and prevented FGFR1-mediated maladaptive subversion of angiocrine factors. Similarly, selective CXCR7 activation in liver sinusoidal endothelial cells abrogated fibrogenesis. Thus, we demonstrate that in response to liver injury, differential recruitment of pro-regenerative CXCR7-Id1 versus pro-fibrotic FGFR1-CXCR4 angiocrine pathways in vascular niche balances regeneration and fibrosis. These results provide a therapeutic roadmap to achieve hepatic regeneration without provoking fibrosis.
Figures
Comment in
-
Sinusoidal endothelial cells direct traffic at the intersection of regeneration and fibrosis.Hepatology. 2014 Aug;60(2):754-6. doi: 10.1002/hep.27116. Epub 2014 May 6. Hepatology. 2014. PMID: 24615996 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
