

Direct, interferon-independent activation of the CXCL10 promoter by NF-κB and interferon regulatory factor 3 during hepatitis C virus infection
- PMID: 24257594
- PMCID: PMC3911583
- DOI: 10.1128/JVI.02007-13
Direct, interferon-independent activation of the CXCL10 promoter by NF-κB and interferon regulatory factor 3 during hepatitis C virus infection
Abstract
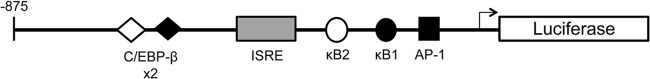
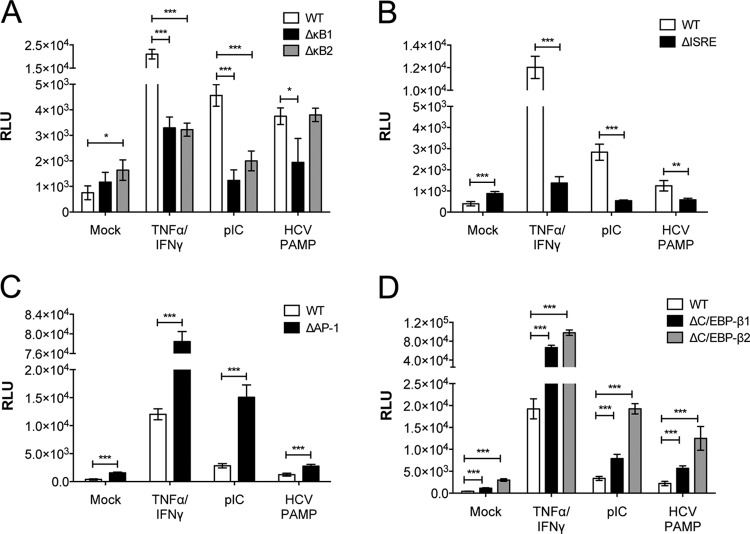
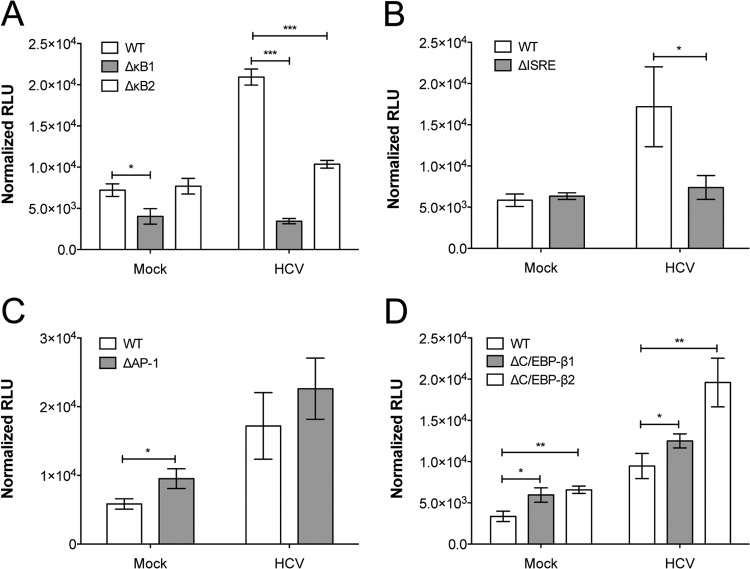
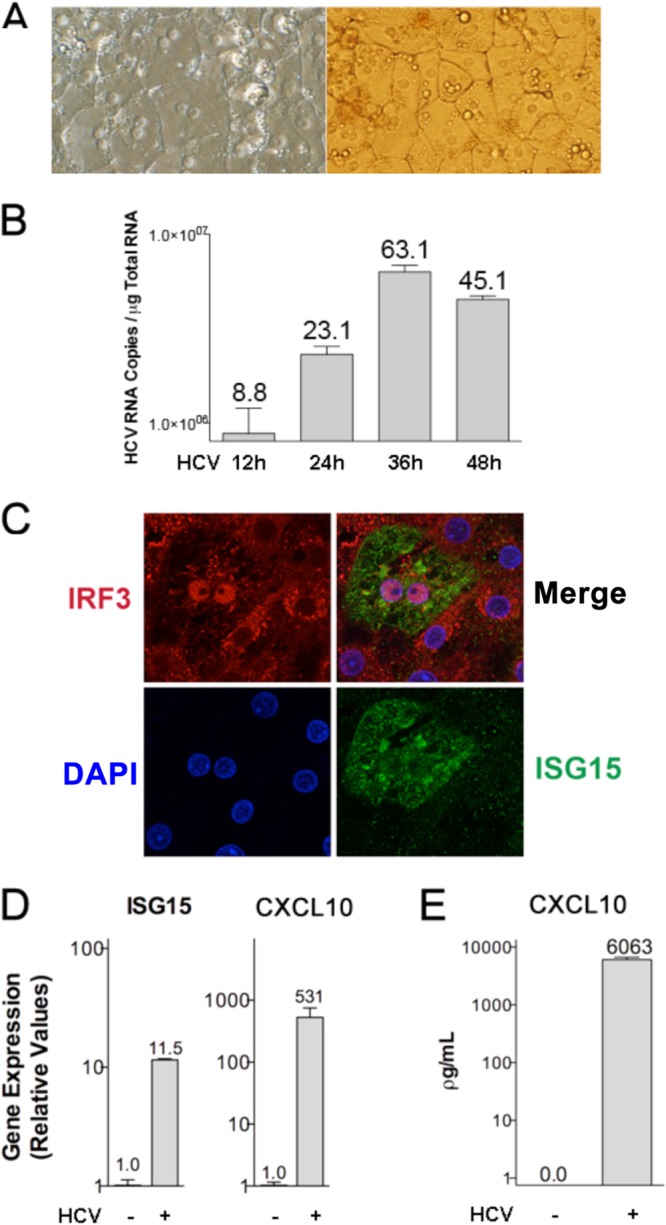
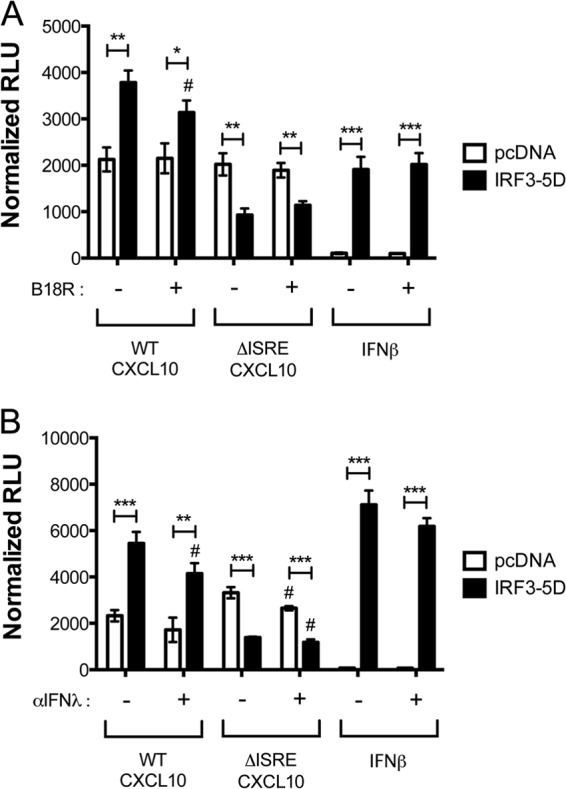
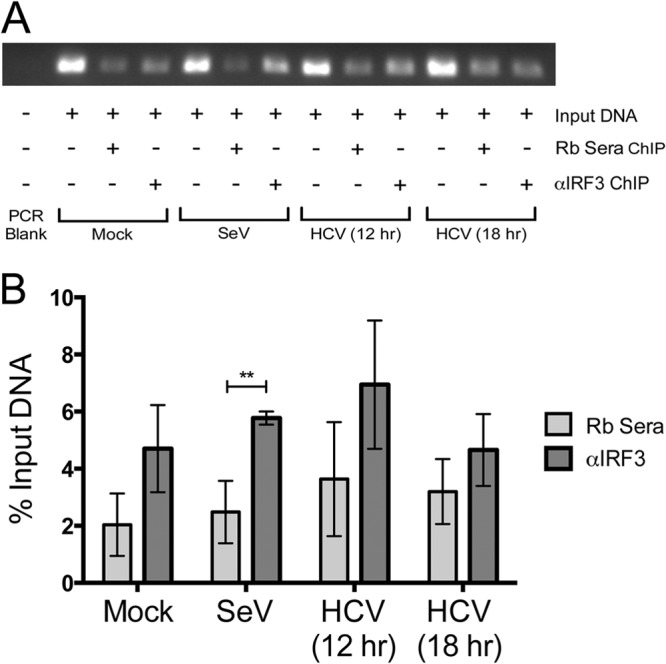
Hepatitis C virus (HCV) infection of hepatocytes leads to transcriptional induction of the chemokine CXCL10, which is considered an interferon (IFN)-stimulated gene. However, we have recently shown that IFNs are not required for CXCL10 induction in hepatocytes during acute HCV infection. Since the CXCL10 promoter contains binding sites for several proinflammatory transcription factors, we investigated the contribution of these factors to CXCL10 transcriptional induction during HCV infection in vitro. Wild-type and mutant CXCL10 promoter-luciferase reporter constructs were used to identify critical sites of transcriptional regulation. The proximal IFN-stimulated response element (ISRE) and NF-κB binding sites positively regulated CXCL10 transcription during HCV infection as well as following exposure to poly(I·C) (a Toll-like receptor 3 [TLR3] stimulus) and 5' poly(U) HCV RNA (a retinoic acid-inducible gene I [RIG-I] stimulus) from two viral genotypes. Conversely, binding sites for AP-1 and CCAAT/enhancer-binding protein β (C/EBP-β) negatively regulated CXCL10 induction in response to TLR3 and RIG-I stimuli, while only C/EBP-β negatively regulated CXCL10 during HCV infection. We also demonstrated that interferon-regulatory factor 3 (IRF3) is transiently recruited to the proximal ISRE during HCV infection and localizes to the nucleus in HCV-infected primary human hepatocytes. Furthermore, IRF3 activated the CXCL10 promoter independently of type I or type III IFN signaling. The data indicate that sensing of HCV infection by RIG-I and TLR3 leads to direct recruitment of NF-κB and IRF3 to the CXCL10 promoter. Our study expands upon current knowledge regarding the mechanisms of CXCL10 induction in hepatocytes and lays the foundation for additional mechanistic studies that further elucidate the combinatorial and synergistic aspects of immune signaling pathways.
Figures
References
-
- Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM. 2012. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55:666–675. 10.1002/hep.24763 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
