

Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies
- PMID: 24257694
- PMCID: PMC3972265
- DOI: 10.1038/ki.2013.450
Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies
Abstract
Rare single-gene disorders cause chronic disease. However, half of the 6000 recessive single gene causes of disease are still unknown. Because recessive disease genes can illuminate, at least in part, disease pathomechanism, their identification offers direct opportunities for improved clinical management and potentially treatment. Rare diseases comprise the majority of chronic kidney disease (CKD) in children but are notoriously difficult to diagnose. Whole-exome resequencing facilitates identification of recessive disease genes. However, its utility is impeded by the large number of genetic variants detected. We here overcome this limitation by combining homozygosity mapping with whole-exome resequencing in 10 sib pairs with a nephronophthisis-related ciliopathy, which represents the most frequent genetic cause of CKD in the first three decades of life. In 7 of 10 sibships with a histologic or ultrasonographic diagnosis of nephronophthisis-related ciliopathy, we detect the causative gene. In six sibships, we identify mutations of known nephronophthisis-related ciliopathy genes, while in two additional sibships we found mutations in the known CKD-causing genes SLC4A1 and AGXT as phenocopies of nephronophthisis-related ciliopathy. Thus, whole-exome resequencing establishes an efficient, noninvasive approach towards early detection and causation-based diagnosis of rare kidney diseases. This approach can be extended to other rare recessive disorders, thereby providing accurate diagnosis and facilitating the study of disease mechanisms.
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures
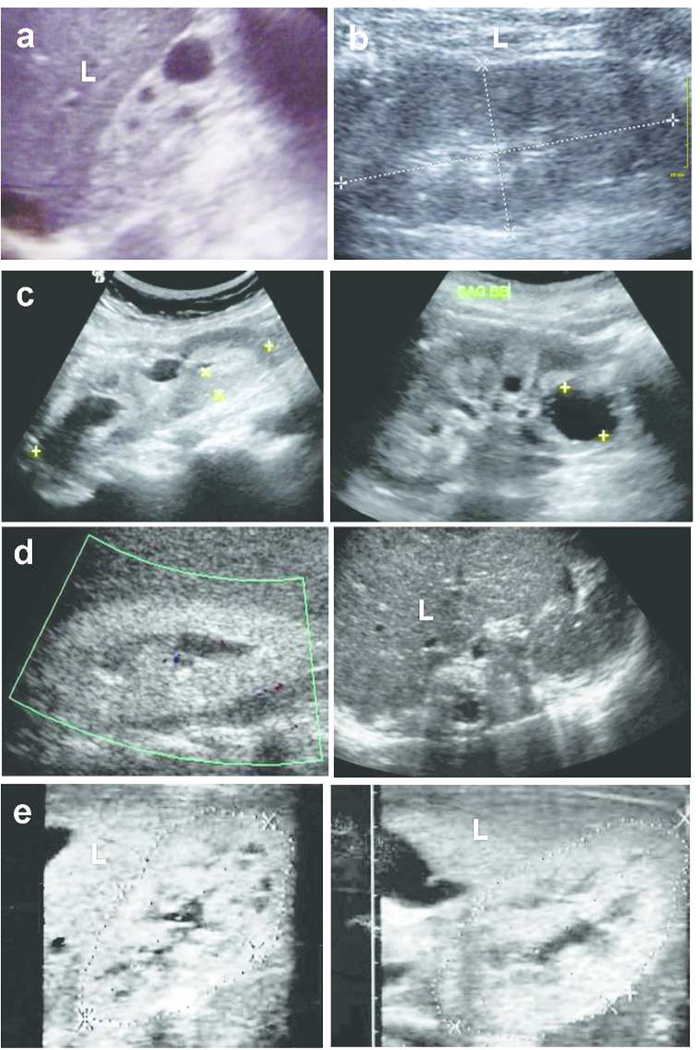
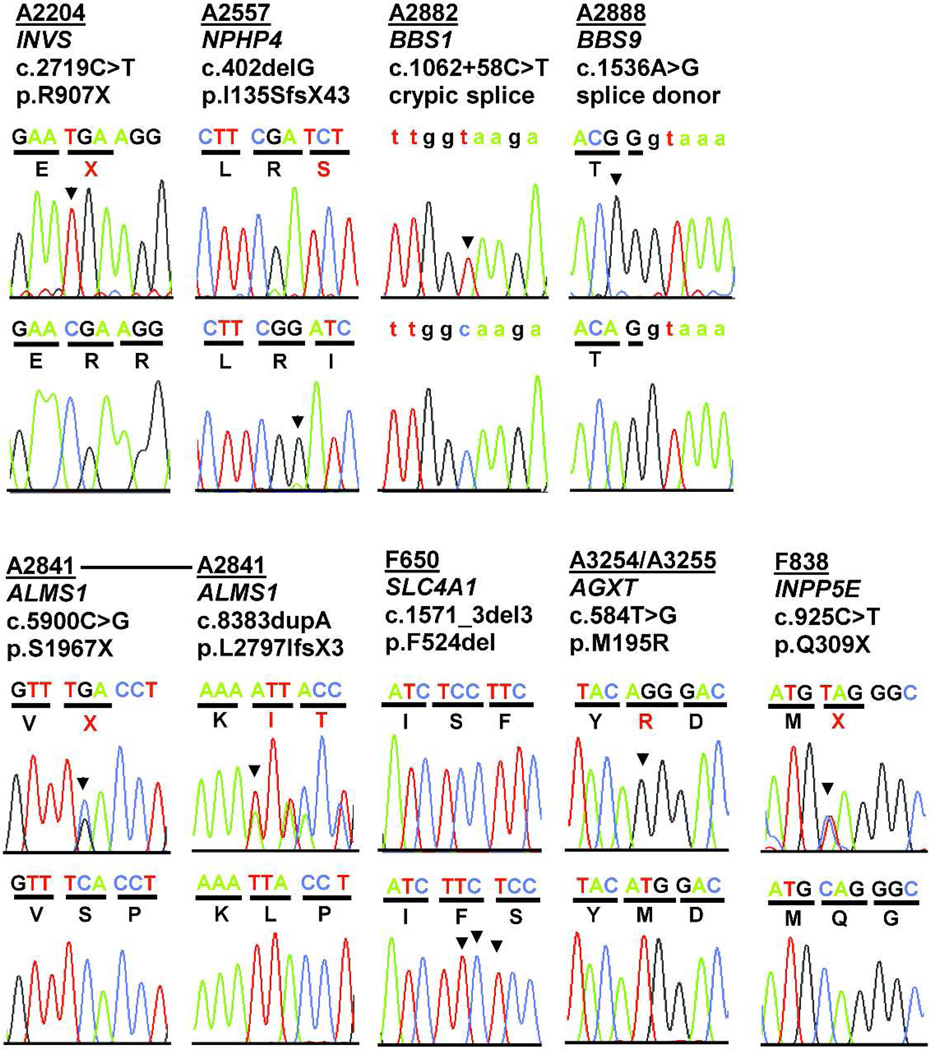
Comment in
-
The challenges and surprises of a definitive molecular genetic diagnosis.Kidney Int. 2014 Apr;85(4):748-9. doi: 10.1038/ki.2013.432. Kidney Int. 2014. PMID: 24682124
References
-
- Smith JM, Stablein DM, Munoz R, et al. Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Pediatr Transplant. 2007;11:366–373. - PubMed
-
- Blowey DL, Querfeld U, Geary D, et al. Ultrasound findings in juvenile nephronophthisis. Pediatr Nephrol. 1996;10:22–24. - PubMed
-
- Zollinger HU, Mihatsch MJ, Edefonti A, et al. Nephronophthisis (medullary cystic disease of the kidney). A study using electron microscopy, immunofluorescence, and a review of the morphological findings. Helv Paediatr Acta. 1980;35:509–530. - PubMed
Publication types
MeSH terms
Grants and funding
- DK1068306/DK/NIDDK NIH HHS/United States
- DK072301/DK/NIDDK NIH HHS/United States
- R01 DK072301/DK/NIDDK NIH HHS/United States
- HD042601/HD/NICHD NIH HHS/United States
- R00 DK091405/DK/NIDDK NIH HHS/United States
- R01 DK075972/DK/NIDDK NIH HHS/United States
- DK064614/DK/NIDDK NIH HHS/United States
- DK075972/DK/NIDDK NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 DK090326/DK/NIDDK NIH HHS/United States
- R01 DK068306/DK/NIDDK NIH HHS/United States
- R01 HD042601/HD/NICHD NIH HHS/United States
- R01 DK064614/DK/NIDDK NIH HHS/United States
- DK1069274/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
