

Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions
- PMID: 24260552
- PMCID: PMC3834214
- DOI: 10.1371/journal.pone.0081162
Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions
Abstract
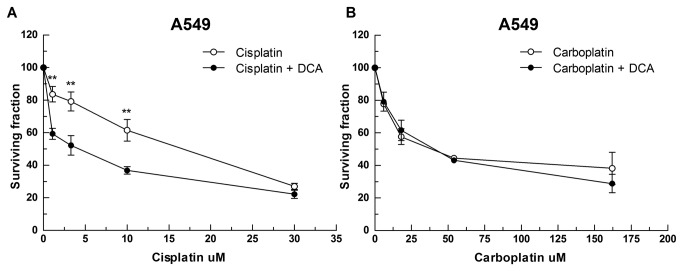
Cisplatin is one of the most effective and widely used anticancer agents for the treatment of several types of tumors. The cytotoxic effect of cisplatin is thought to be mediated primarily by the generation of nuclear DNA adducts, which, if not repaired, cause cell death as a consequence of DNA replication and transcription blockage. However, the ability of cisplatin to induce nuclear DNA (nDNA) damage per se is not sufficient to explain its high degree of effectiveness nor the toxic effects exerted on normal, post-mitotic tissues. Oxidative damage has been observed in vivo following exposure to cisplatin in several tissues, suggesting a role for oxidative stress in the pathogenesis of cisplatin-induced dose-limiting toxicities. However, the mechanism of cisplatin-induced generation of ROS and their contribution to cisplatin cytotoxicity in normal and cancer cells is still poorly understood. By employing a panel of normal and cancer cell lines and the budding yeast Saccharomyces cerevisiae as model system, we show that exposure to cisplatin induces a mitochondrial-dependent ROS response that significantly enhances the cytotoxic effect caused by nDNA damage. ROS generation is independent of the amount of cisplatin-induced nDNA damage and occurs in mitochondria as a consequence of protein synthesis impairment. The contribution of cisplatin-induced mitochondrial dysfunction in determining its cytotoxic effect varies among cells and depends on mitochondrial redox status, mitochondrial DNA integrity and bioenergetic function. Thus, by manipulating these cellular parameters, we were able to enhance cisplatin cytotoxicity in cancer cells. This study provides a new mechanistic insight into cisplatin-induced cell killing and may lead to the design of novel therapeutic strategies to improve anticancer drug efficacy.
Conflict of interest statement
Figures
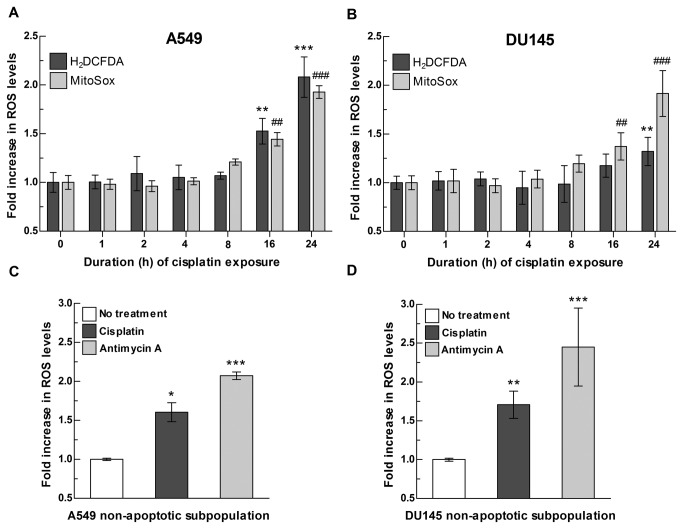
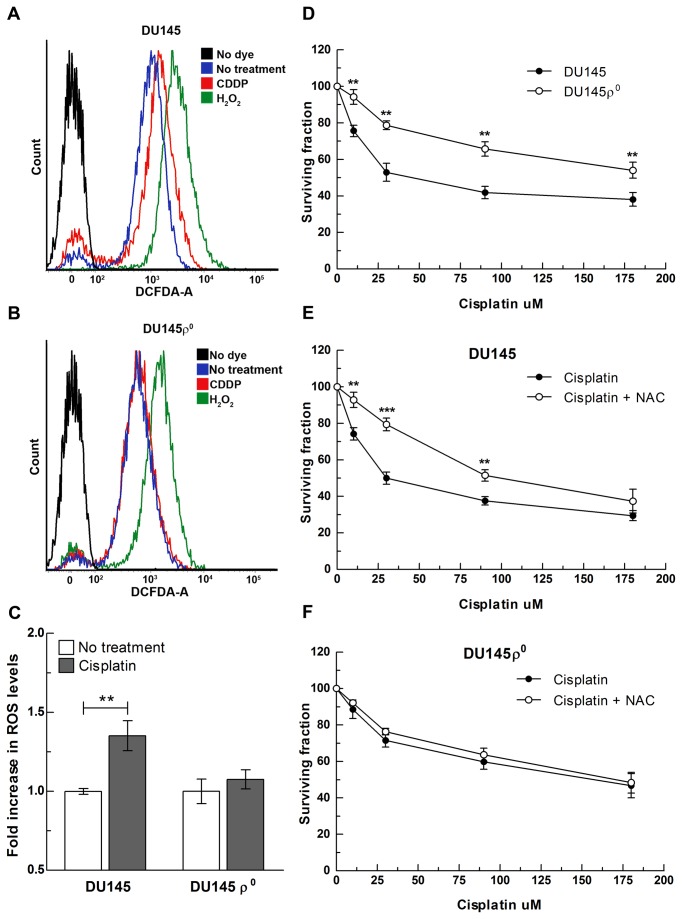
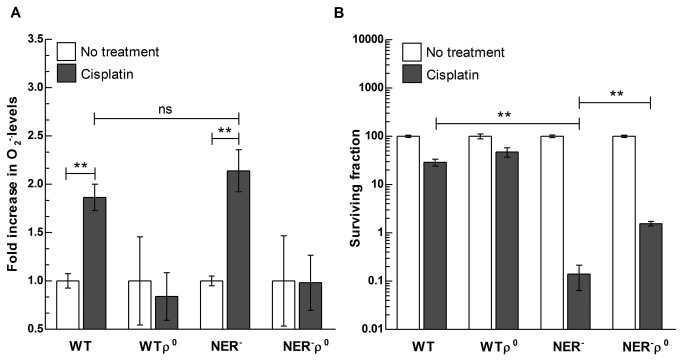

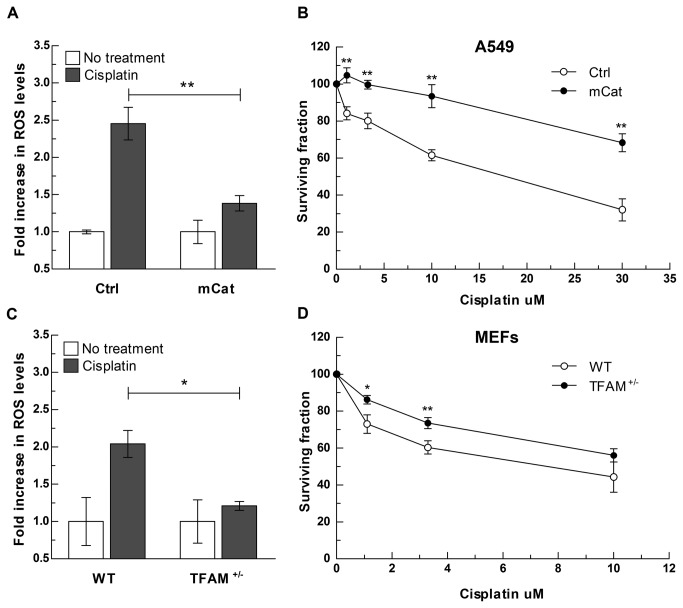

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
