

Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia
- PMID: 24265505
- PMCID: PMC3912412
- DOI: 10.1101/gr.152132.112
Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia
Abstract
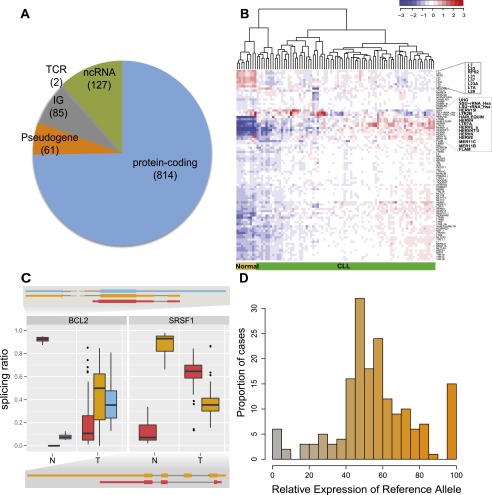
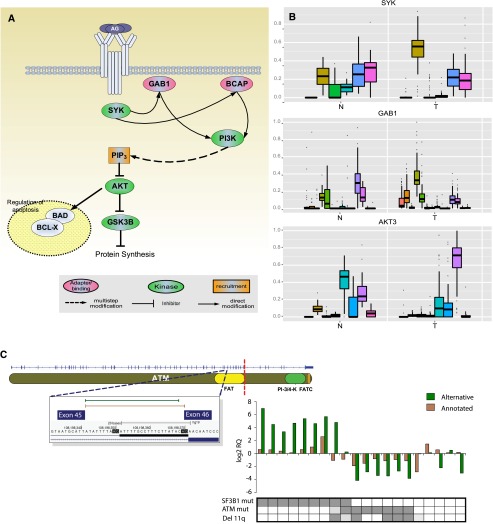
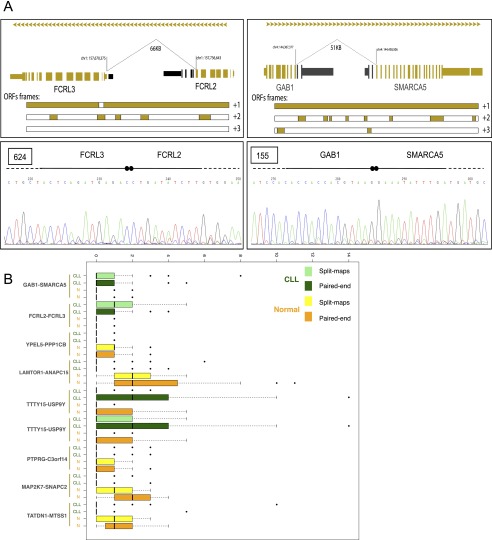
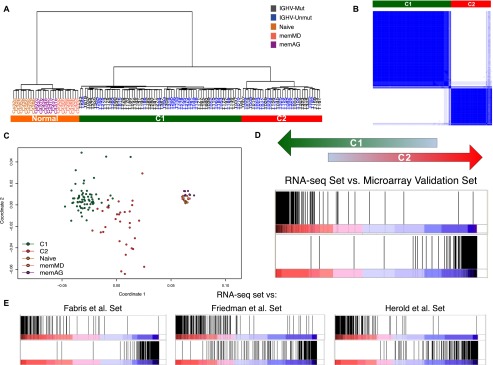
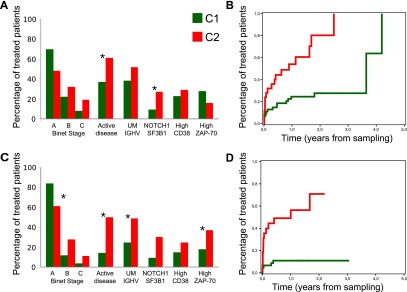
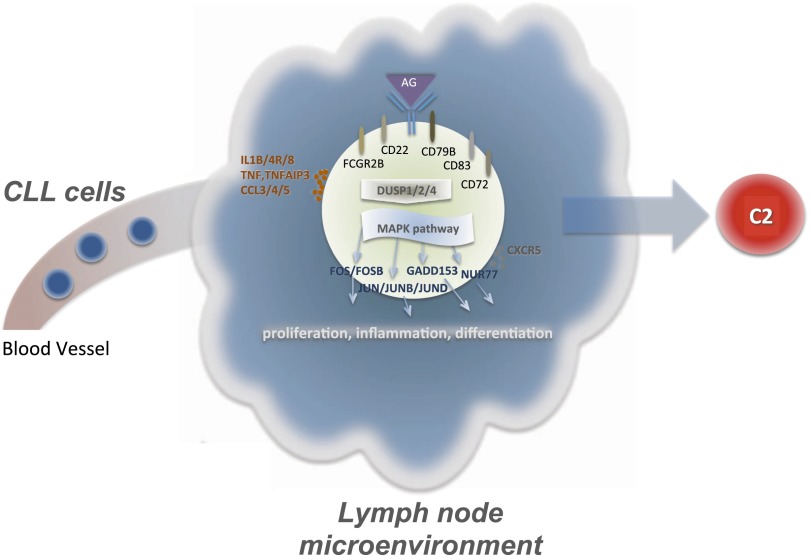
Chronic lymphocytic leukemia (CLL) has heterogeneous clinical and biological behavior. Whole-genome and -exome sequencing has contributed to the characterization of the mutational spectrum of the disease, but the underlying transcriptional profile is still poorly understood. We have performed deep RNA sequencing in different subpopulations of normal B-lymphocytes and CLL cells from a cohort of 98 patients, and characterized the CLL transcriptional landscape with unprecedented resolution. We detected thousands of transcriptional elements differentially expressed between the CLL and normal B cells, including protein-coding genes, noncoding RNAs, and pseudogenes. Transposable elements are globally derepressed in CLL cells. In addition, two thousand genes-most of which are not differentially expressed-exhibit CLL-specific splicing patterns. Genes involved in metabolic pathways showed higher expression in CLL, while genes related to spliceosome, proteasome, and ribosome were among the most down-regulated in CLL. Clustering of the CLL samples according to RNA-seq derived gene expression levels unveiled two robust molecular subgroups, C1 and C2. C1/C2 subgroups and the mutational status of the immunoglobulin heavy variable (IGHV) region were the only independent variables in predicting time to treatment in a multivariate analysis with main clinico-biological features. This subdivision was validated in an independent cohort of patients monitored through DNA microarrays. Further analysis shows that B-cell receptor (BCR) activation in the microenvironment of the lymph node may be at the origin of the C1/C2 differences.
Figures
References
-
- Antonov AV 2011. BioProfiling.de: Analytical web portal for high-throughput cell biology. Nucleic Acids Res 39: W323–W327 - PMC - PubMed
-
- Buchner M, Fuchs S, Prinz G, Pfeifer D, Bartholome K, Burger M, Chevalier N, Vallat L, Timmer J, Gribben JG, et al. 2009. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res 69: 5424–5432 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous