

Reverse causal reasoning: applying qualitative causal knowledge to the interpretation of high-throughput data
- PMID: 24266983
- PMCID: PMC4222496
- DOI: 10.1186/1471-2105-14-340
Reverse causal reasoning: applying qualitative causal knowledge to the interpretation of high-throughput data
Abstract
Background: Gene expression profiling and other genome-scale measurement technologies provide comprehensive information about molecular changes resulting from a chemical or genetic perturbation, or disease state. A critical challenge is the development of methods to interpret these large-scale data sets to identify specific biological mechanisms that can provide experimentally verifiable hypotheses and lead to the understanding of disease and drug action.
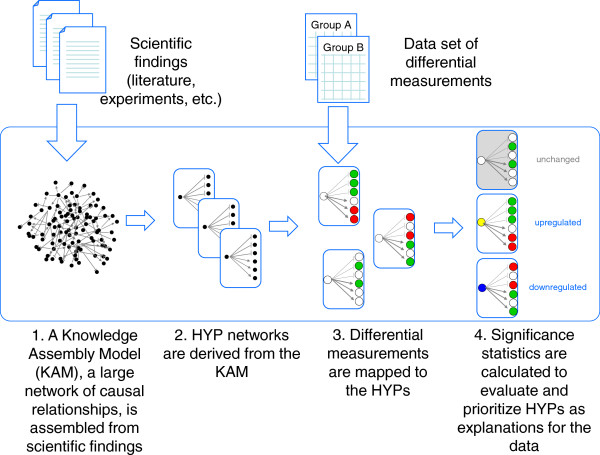
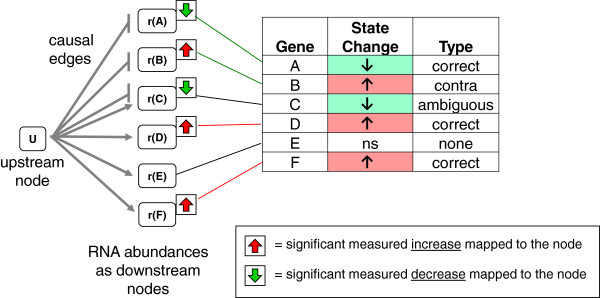
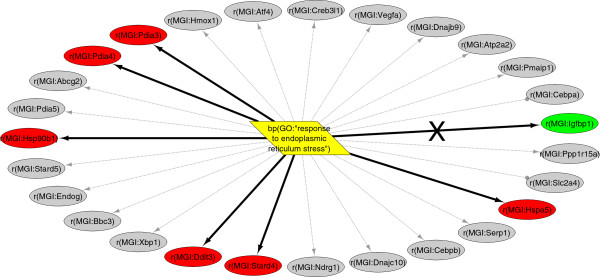
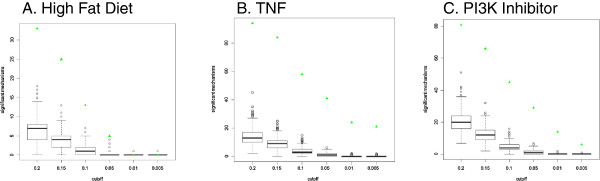
Results: We present a detailed description of Reverse Causal Reasoning (RCR), a reverse engineering methodology to infer mechanistic hypotheses from molecular profiling data. This methodology requires prior knowledge in the form of small networks that causally link a key upstream controller node representing a biological mechanism to downstream measurable quantities. These small directed networks are generated from a knowledge base of literature-curated qualitative biological cause-and-effect relationships expressed as a network. The small mechanism networks are evaluated as hypotheses to explain observed differential measurements. We provide a simple implementation of this methodology, Whistle, specifically geared towards the analysis of gene expression data and using prior knowledge expressed in Biological Expression Language (BEL). We present the Whistle analyses for three transcriptomic data sets using a publically available knowledge base. The mechanisms inferred by Whistle are consistent with the expected biology for each data set.
Conclusions: Reverse Causal Reasoning yields mechanistic insights to the interpretation of gene expression profiling data that are distinct from and complementary to the results of analyses using ontology or pathway gene sets. This reverse engineering algorithm provides an evidence-driven approach to the development of models of disease, drug action, and drug toxicity.
Figures
References
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
