

MMDiff: quantitative testing for shape changes in ChIP-Seq data sets
- PMID: 24267901
- PMCID: PMC4008153
- DOI: 10.1186/1471-2164-14-826
MMDiff: quantitative testing for shape changes in ChIP-Seq data sets
Abstract
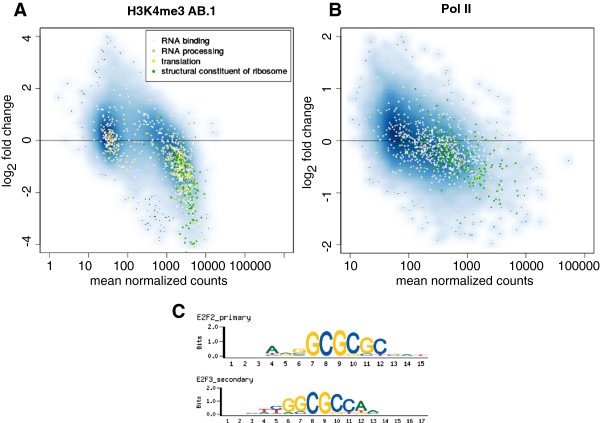
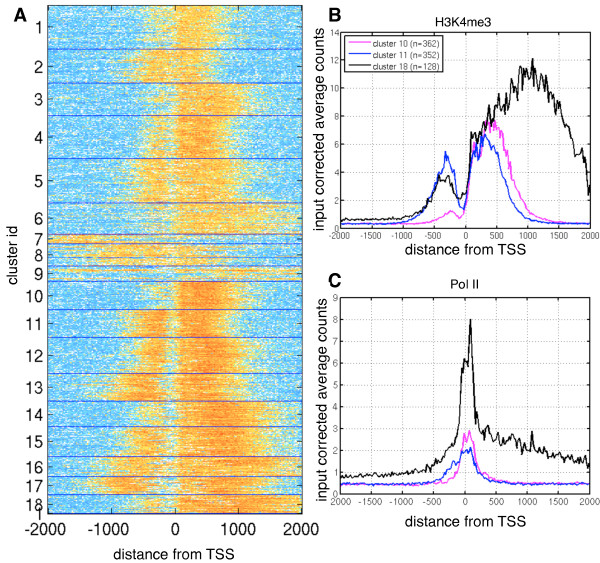
Background: Cell-specific gene expression is controlled by epigenetic modifications and transcription factor binding. While genome-wide maps for these protein-DNA interactions have become widely available, quantitative comparison of the resulting ChIP-Seq data sets remains challenging. Current approaches to detect differentially bound or modified regions are mainly borrowed from RNA-Seq data analysis, thus focusing on total counts of fragments mapped to a region, ignoring any information encoded in the shape of the peaks.
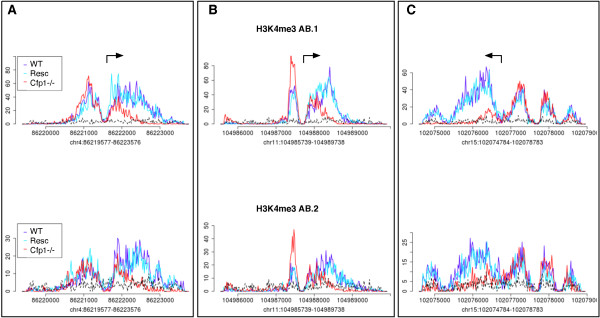
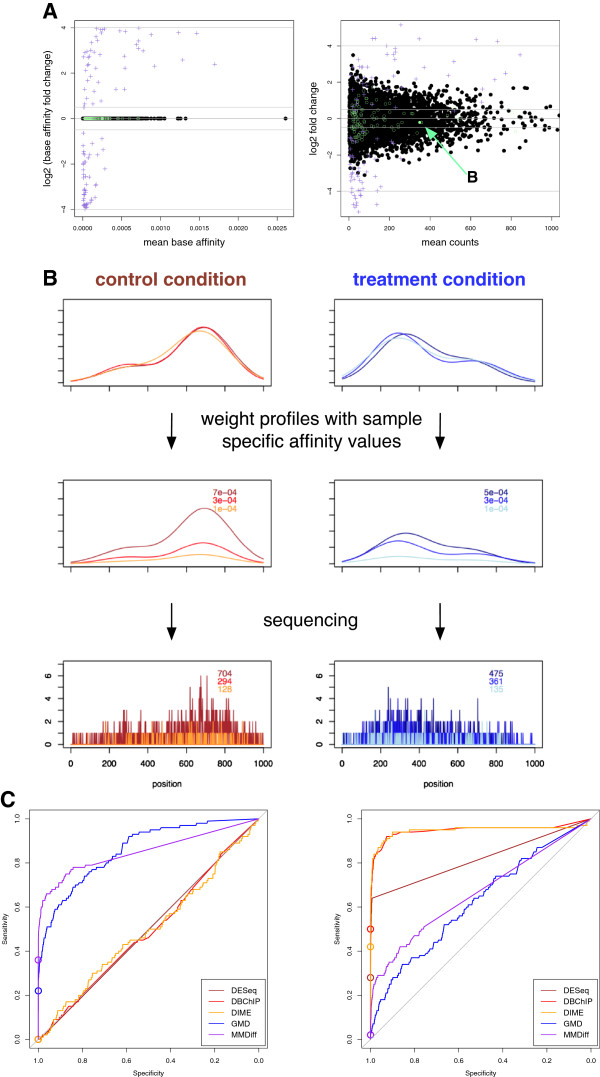
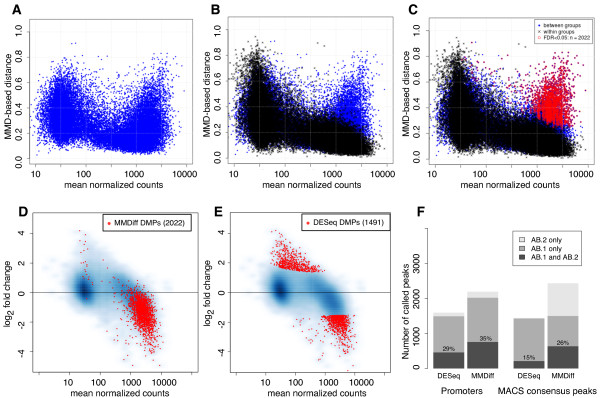
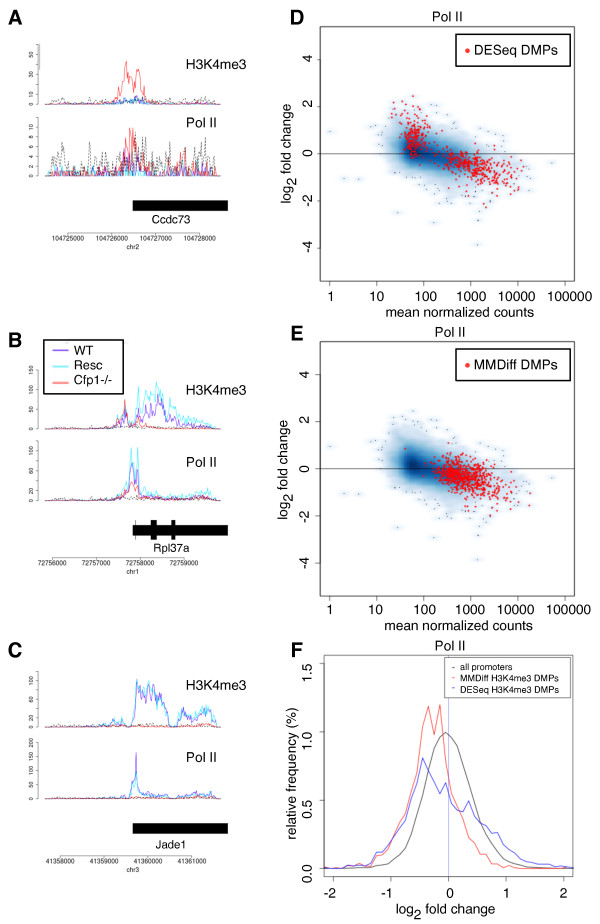
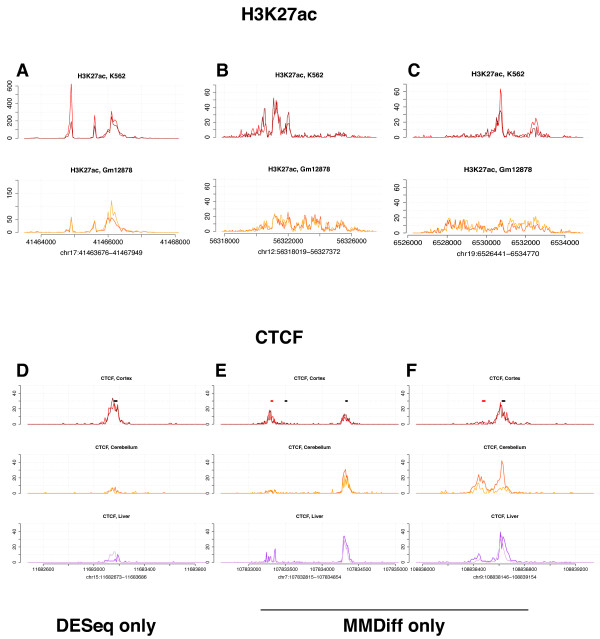
Results: Here, we present MMDiff, a robust, broadly applicable method for detecting differences between sequence count data sets. Based on quantifying shape changes in signal profiles, it overcomes challenges imposed by the highly structured nature of the data and the paucity of replicates.We first use a simulated data set to compare the performance of MMDiff with results obtained by four alternative methods. We demonstrate that MMDiff excels when peak profiles change between samples. We next use MMDiff to re-analyse a recent data set of the histone modification H3K4me3 elucidating the establishment of this prominent epigenomic marker. Our empirical analysis shows that the method yields reproducible results across experiments, and is able to detect functional important changes in histone modifications. To further explore the broader applicability of MMDiff, we apply it to two ENCODE data sets: one investigating the histone modification H3K27ac and one measuring the genome-wide binding of the transcription factor CTCF. In both cases, MMDiff proves to be complementary to count-based methods. In addition, we can show that MMDiff is capable of directly detecting changes of homotypic binding events at neighbouring binding sites. MMDiff is readily available as a Bioconductor package.
Conclusions: Our results demonstrate that higher order features of ChIP-Seq peaks carry relevant and often complementary information to total counts, and hence are important in assessing differential histone modifications and transcription factor binding. We have developed a new computational method, MMDiff, that is capable of exploring these features and therefore closes an existing gap in the analysis of ChIP-Seq data sets.
Figures
References
-
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, Talianidis I, Flicek P, Odom DT. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;14(5981):1036–1040. doi: 10.1126/science.1186176. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
