

β-Catenin serves as a clutch between low and high intercellular E-cadherin bond strengths
- PMID: 24268141
- PMCID: PMC3838741
- DOI: 10.1016/j.bpj.2013.09.044
β-Catenin serves as a clutch between low and high intercellular E-cadherin bond strengths
Abstract
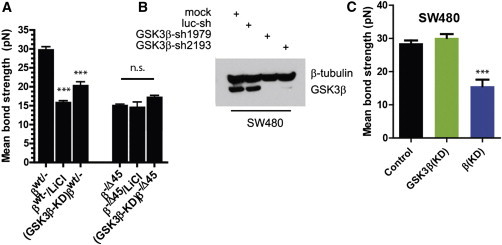
A wide range of invasive pathological outcomes originate from the loss of epithelial phenotype and involve either loss of function or downregulation of transmembrane adhesive receptor complexes, including Ecadherin (Ecad) and binding partners β-catenin and α-catenin at adherens junctions. Cellular pathways regulating wild-type β-catenin level, or direct mutations in β-catenin that affect the turnover of the protein have been shown to contribute to cancer development, through induction of uncontrolled proliferation of transformed tumor cells, particularly in colon cancer. Using single-molecule force spectroscopy, we show that depletion of β-catenin or the prominent cancer-related S45 deletion mutation in β-catenin present in human colon cancers both weaken tumor intercellular Ecad/Ecad bond strength and diminishes the capacity of specific extracellular matrix proteins-including collagen I, collagen IV, and laminin V-to modulate intercellular Ecad/Ecad bond strength through α-catenin and the kinase activity of glycogen synthase kinase 3 (GSK-3β). Thus, in addition to regulating tumor cell proliferation, cancer-related mutations in β-catenin can influence tumor progression by weakening the adhesion of tumor cells to one another through reduced individual Ecad/Ecad bond strength and cellular adhesion to specific components of the extracellular matrix and the basement membrane.
Copyright © 2013 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
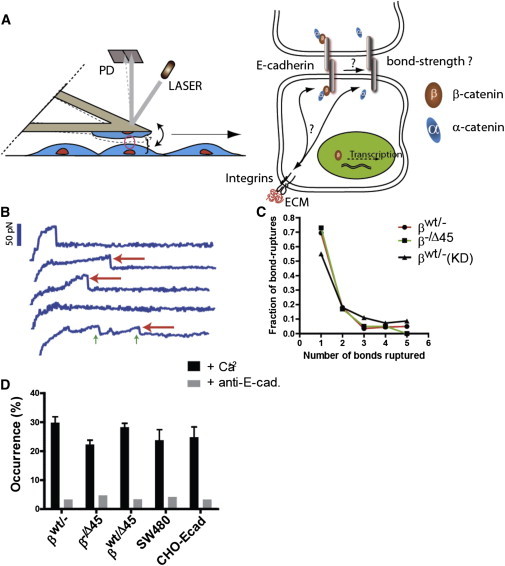
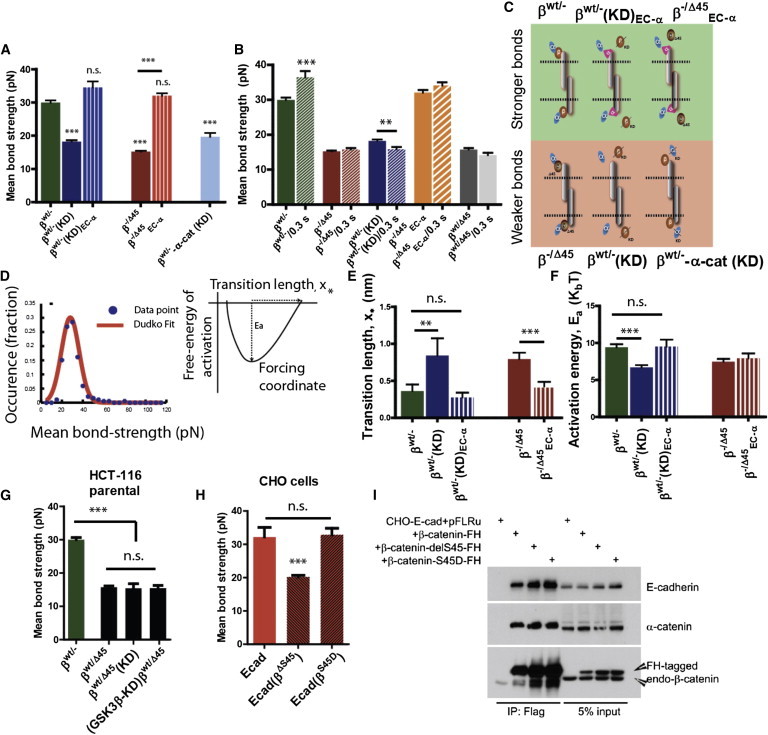
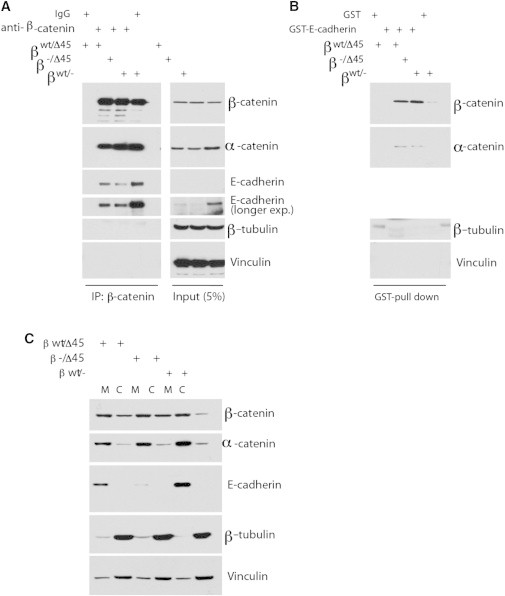
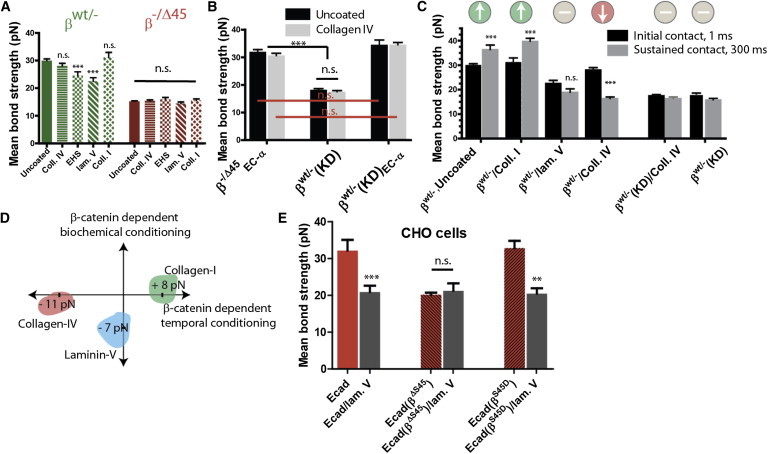
References
-
- Chou J.J., Gaemers S., Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR. 2001;21:377–382. - PubMed
-
- Zimrin A.B., Villeponteau B., Maciag T. Models of in vitro angiogenesis: endothelial cell differentiation on fibrin but not matrigel is transcriptionally dependent. Biochem. Biophys. Res. Commun. 1995;213:630–638. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
