

High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis
- PMID: 24269006
- PMCID: PMC3956118
- DOI: 10.1016/j.cell.2013.10.047
High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis
Abstract
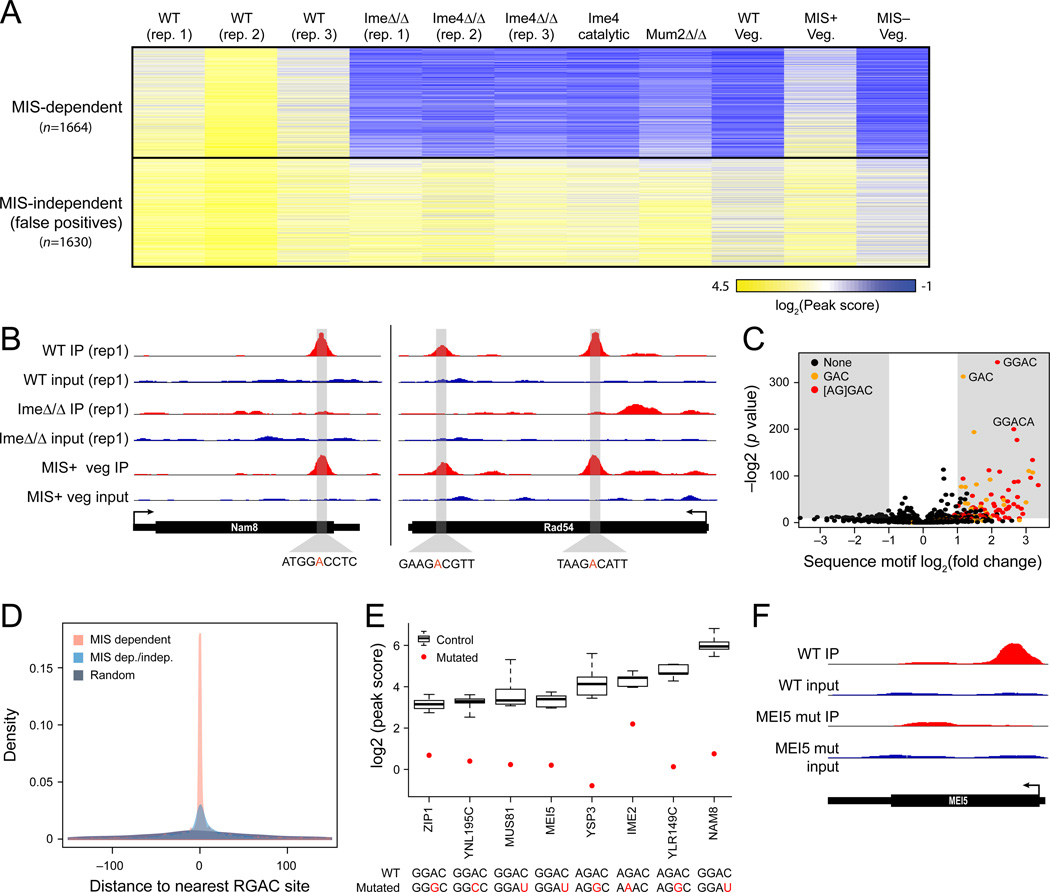
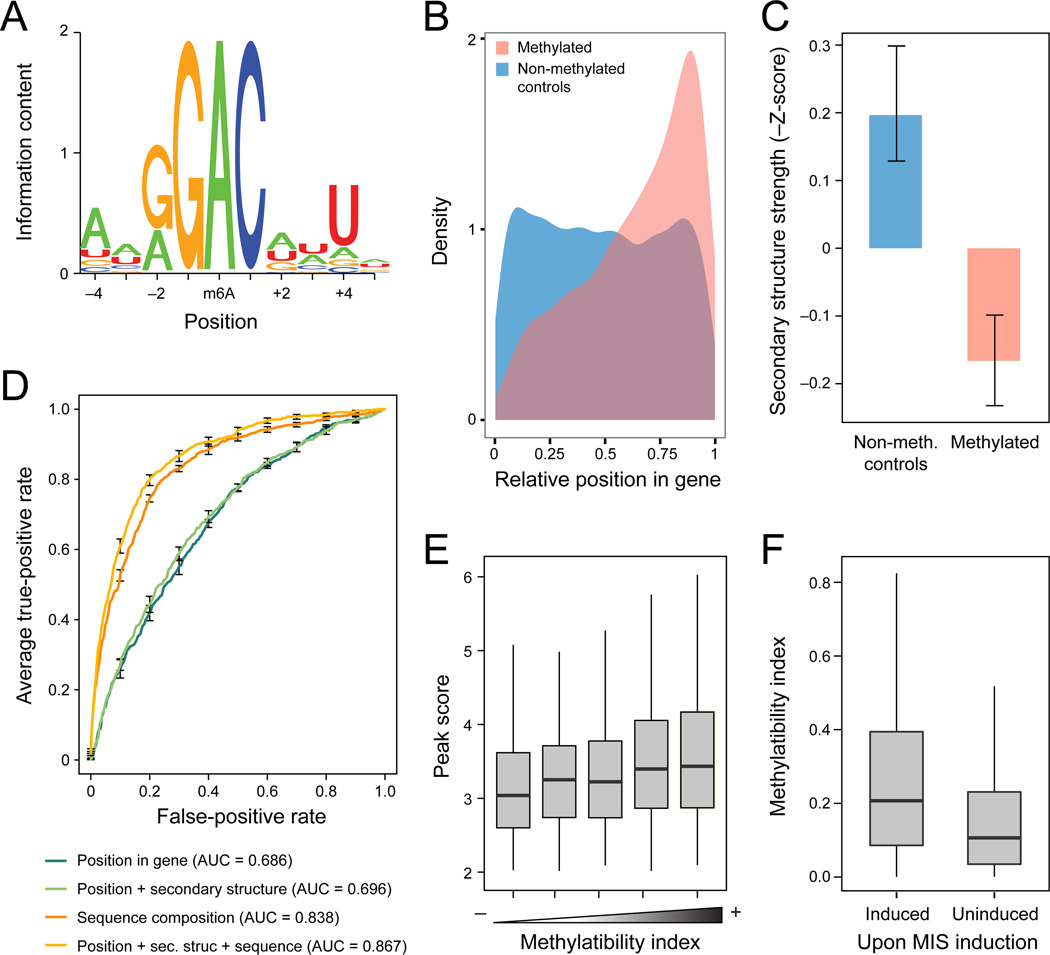
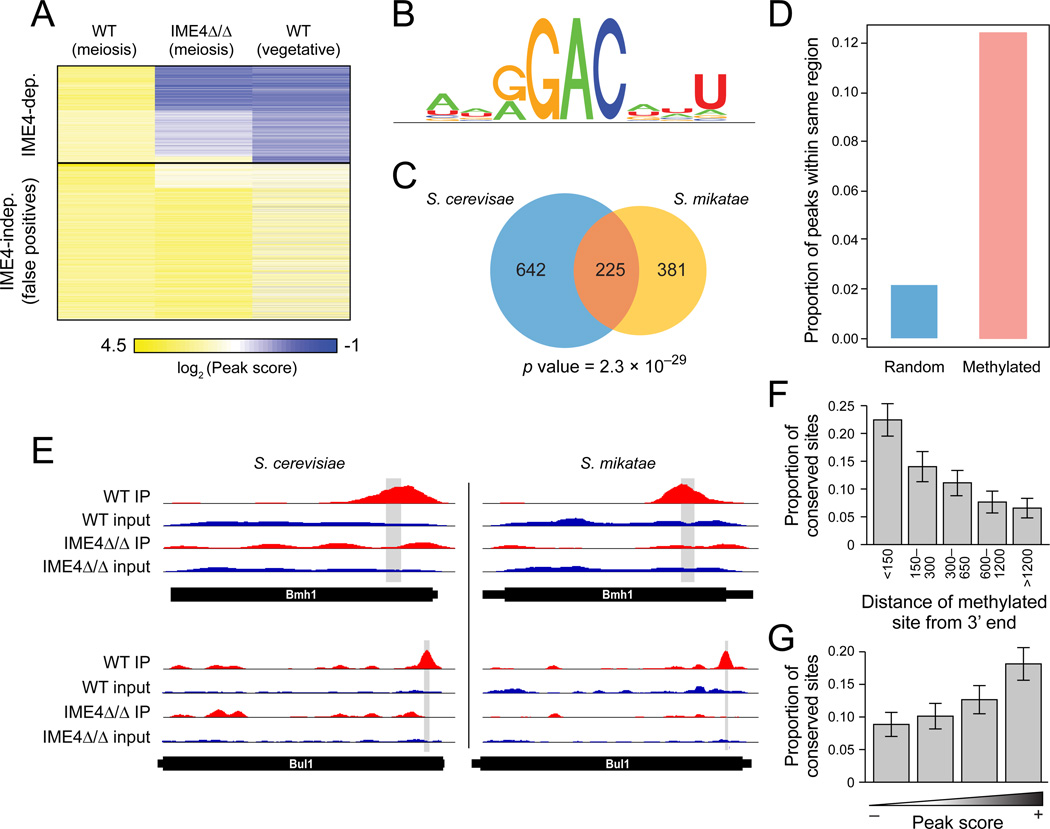
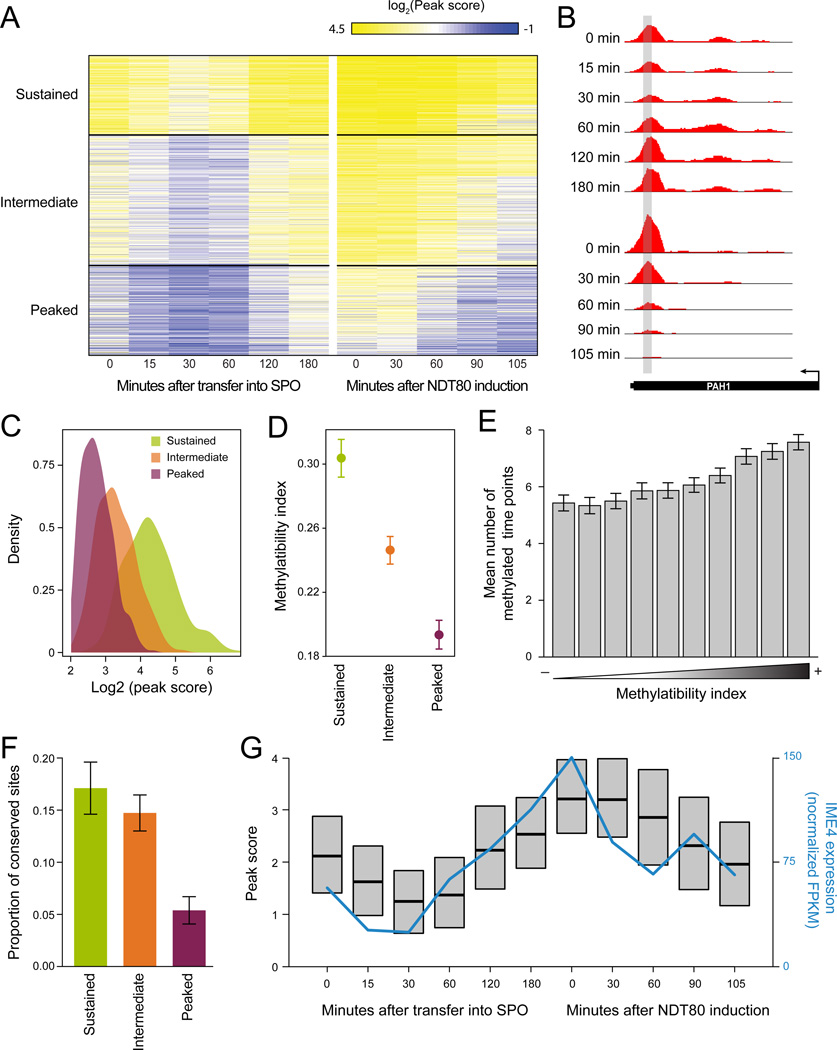
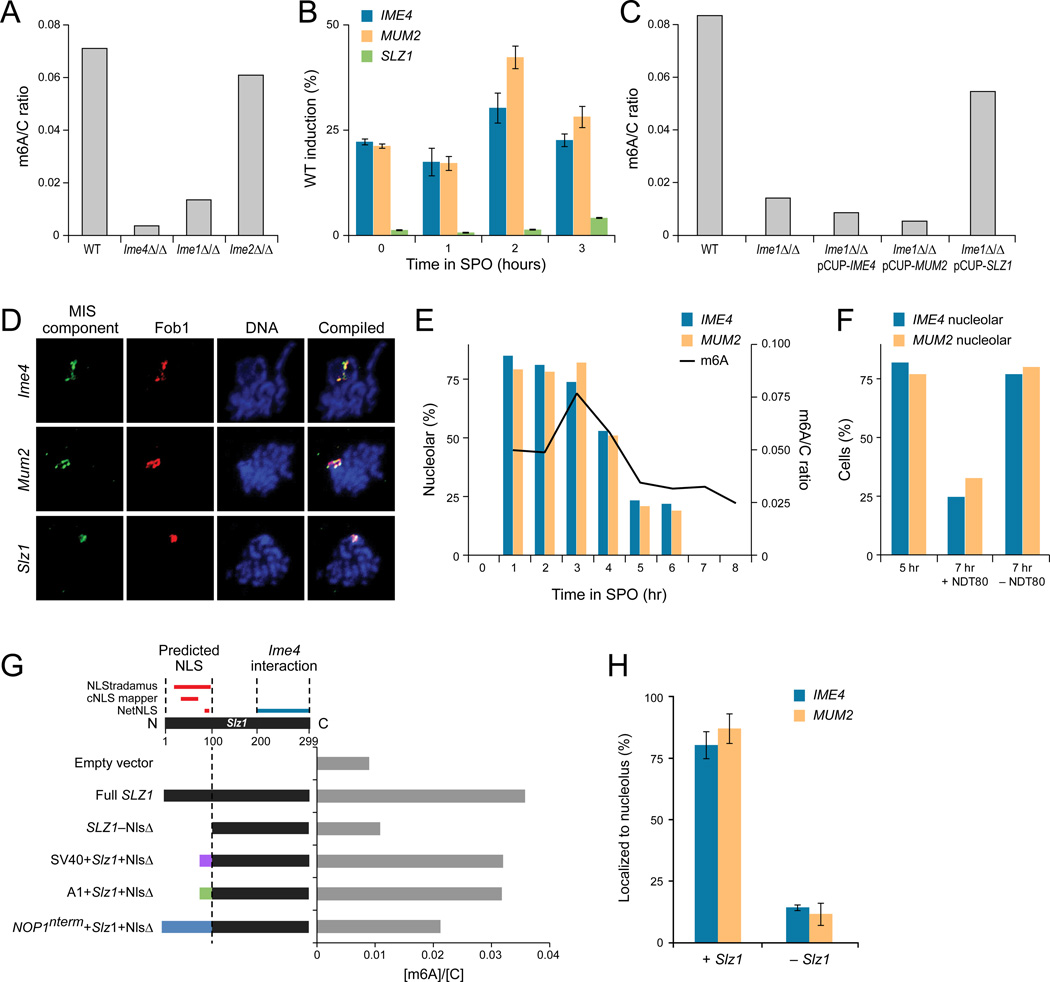
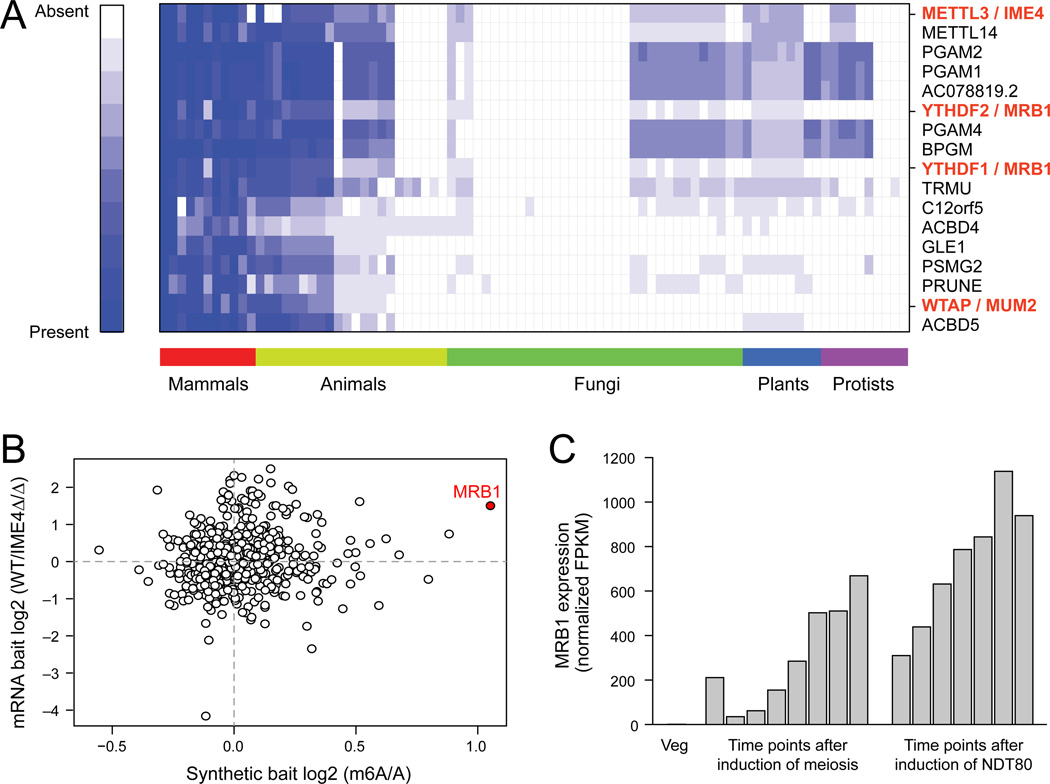
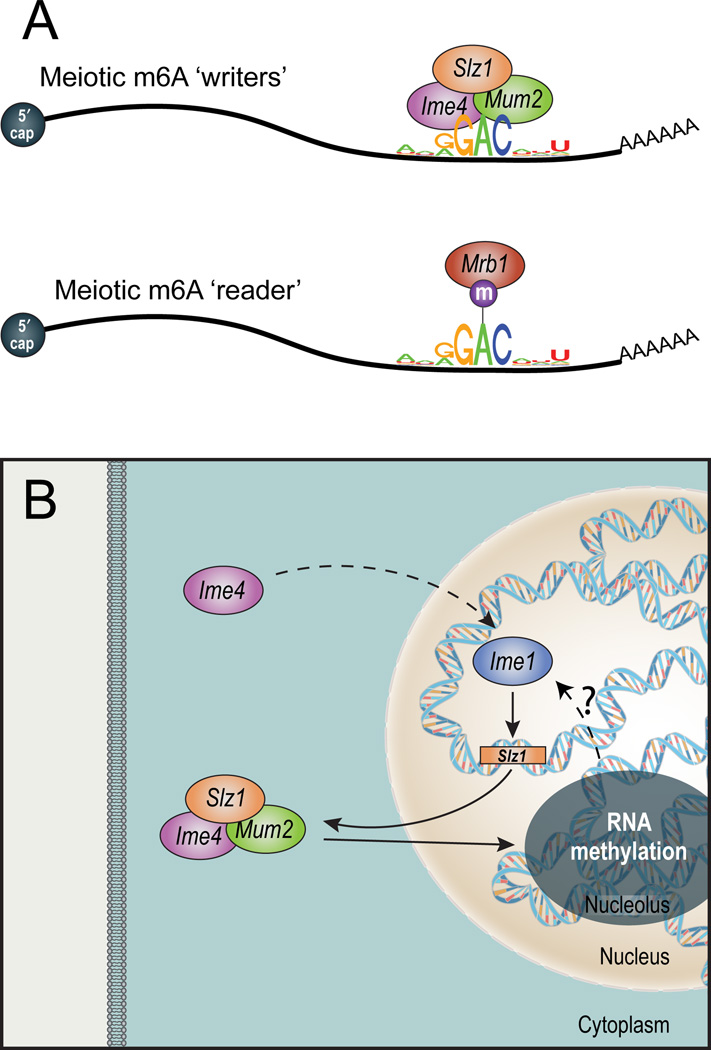
N(6)-methyladenosine (m(6)A) is the most ubiquitous mRNA base modification, but little is known about its precise location, temporal dynamics, and regulation. Here, we generated genomic maps of m(6)A sites in meiotic yeast transcripts at nearly single-nucleotide resolution, identifying 1,308 putatively methylated sites within 1,183 transcripts. We validated eight out of eight methylation sites in different genes with direct genetic analysis, demonstrated that methylated sites are significantly conserved in a related species, and built a model that predicts methylated sites directly from sequence. Sites vary in their methylation profiles along a dense meiotic time course and are regulated both locally, via predictable methylatability of each site, and globally, through the core meiotic circuitry. The methyltransferase complex components localize to the yeast nucleolus, and this localization is essential for mRNA methylation. Our data illuminate a conserved, dynamically regulated methylation program in yeast meiosis and provide an important resource for studying the function of this epitranscriptomic modification.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nature reviews Molecular cell biology. 2007;8:574–585. - PubMed
-
- Bokar JA. Grosjean Henri., editor. The biosynthesis and functional roles of methylated nucleosides in eukaryotic mRNA. Fine-Tuning of RNA Functions by Modification and Editing. 2005;12:141–177.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
