

Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response
- PMID: 24269668
- PMCID: PMC3947349
- DOI: 10.1016/j.it.2013.09.001
Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response
Abstract
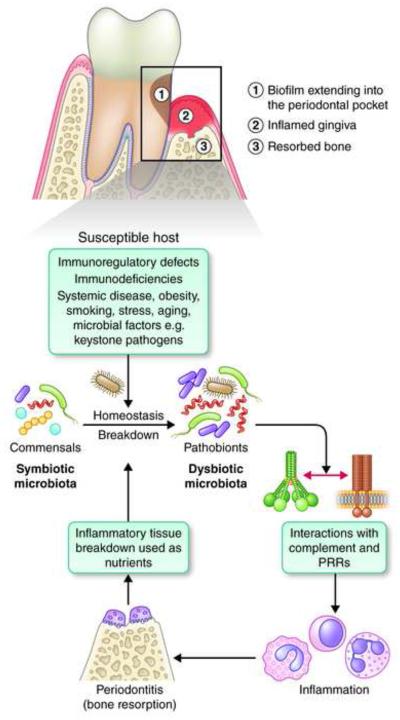
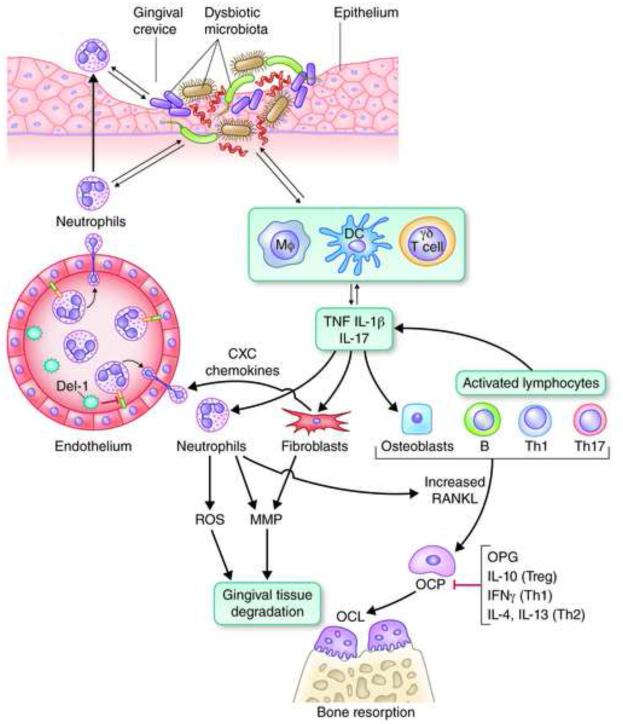
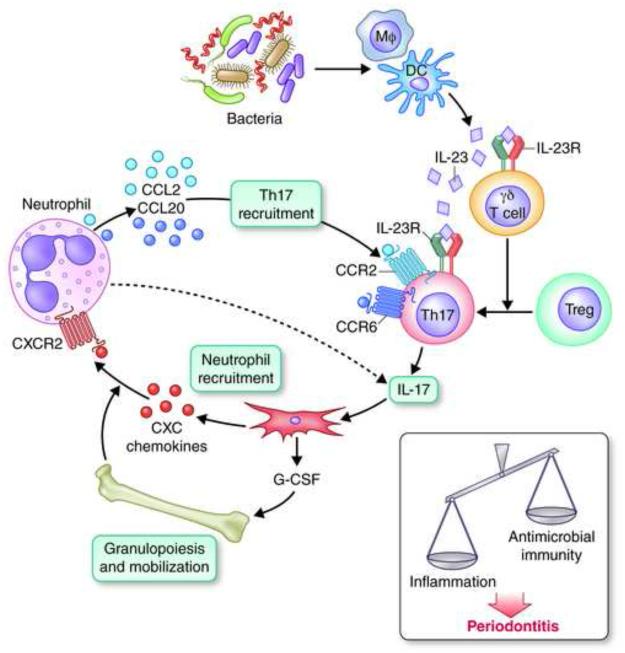
Recent studies have uncovered novel mechanisms underlying the breakdown of periodontal host-microbe homeostasis, which can precipitate dysbiosis and periodontitis in susceptible hosts. Dysbiotic microbial communities of keystone pathogens and pathobionts are thought to exhibit synergistic virulence whereby not only can they endure the host response but can also thrive by exploiting tissue-destructive inflammation, which fuels a self-feeding cycle of escalating dysbiosis and inflammatory bone loss, potentially leading to tooth loss and systemic complications. Here, I discuss new paradigms in our understanding of periodontitis, which may shed light into other polymicrobial inflammatory disorders. In addition, I highlight gaps in knowledge required for an integrated picture of the interplay between microbes and innate and adaptive immune elements that initiate and propagate chronic periodontal inflammation.
Keywords: dysbiosis; inflammation; keystone pathogen; pathobiont; periodontitis.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
References
-
- Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010; 8:481–490. - PubMed
-
- Eke PI, et al. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res. 2012; 91:914–920. - PubMed
-
- Genco RJ, Van Dyke TE. Prevention: Reducing the risk of CVD in patients with periodontitis. Nat Rev Cardiol. 2010; 7:479–480. - PubMed
-
- Lundberg K, et al. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol. 2010; 6:727–730. - PubMed
-
- Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011; 7:738–748. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
