

Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration
- PMID: 24270424
- PMCID: PMC3859381
- DOI: 10.1172/JCI70317
Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration
Abstract
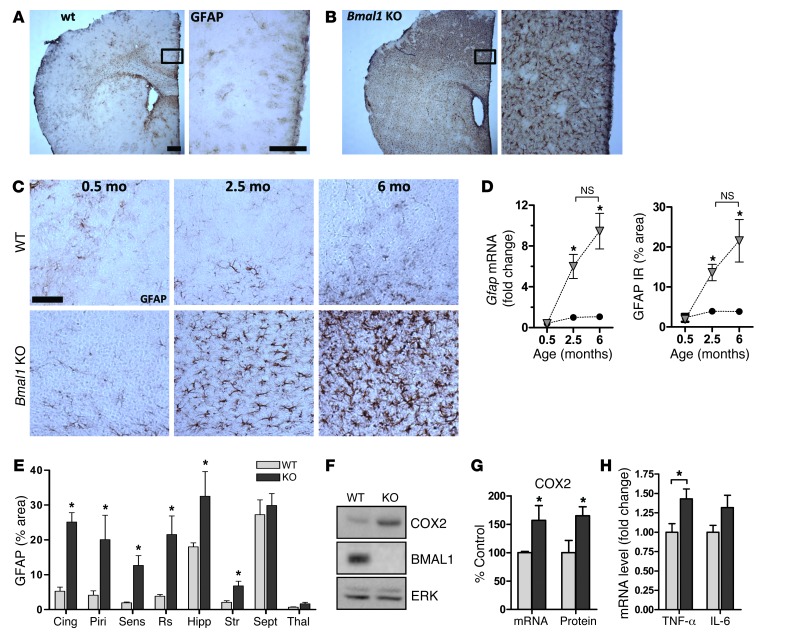
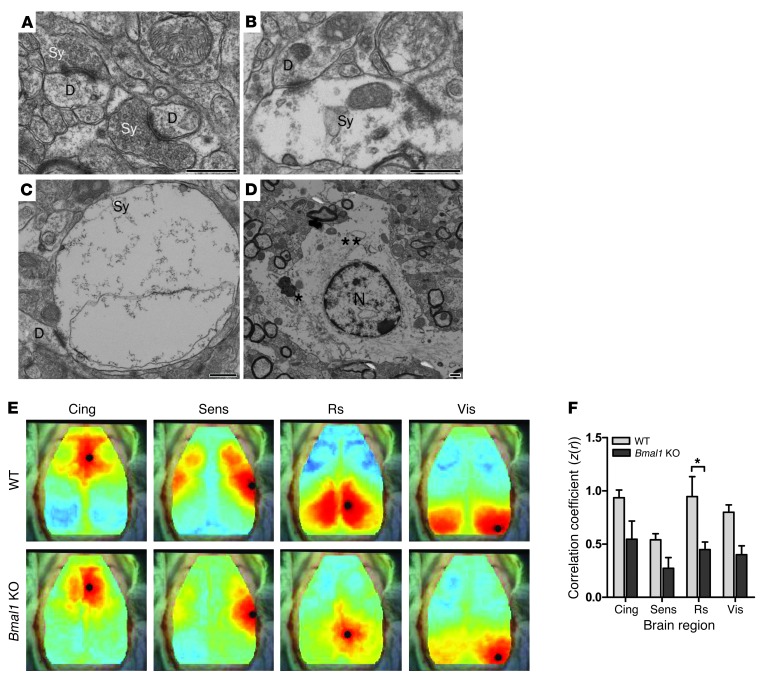
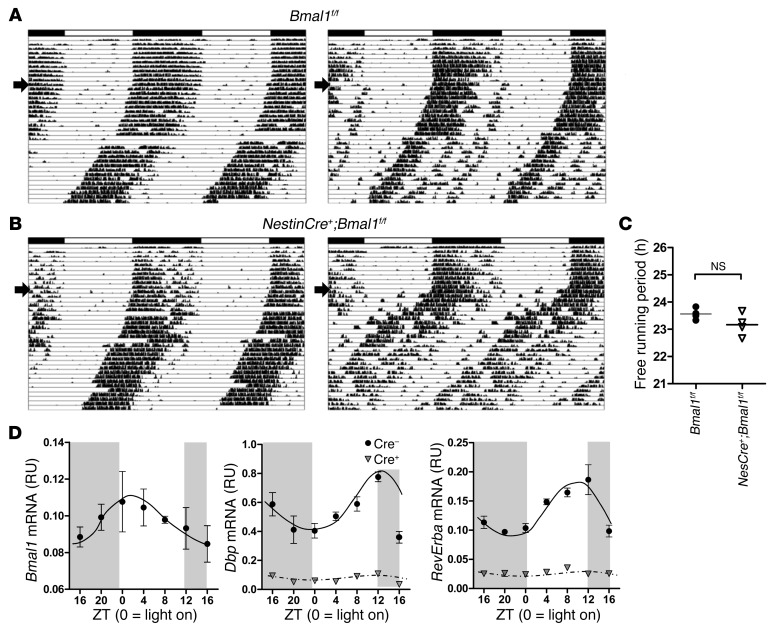
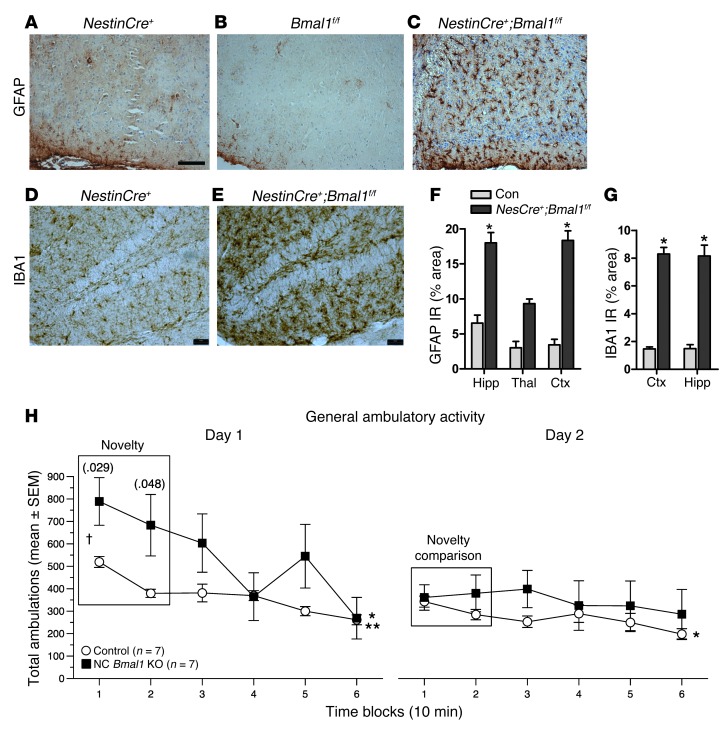
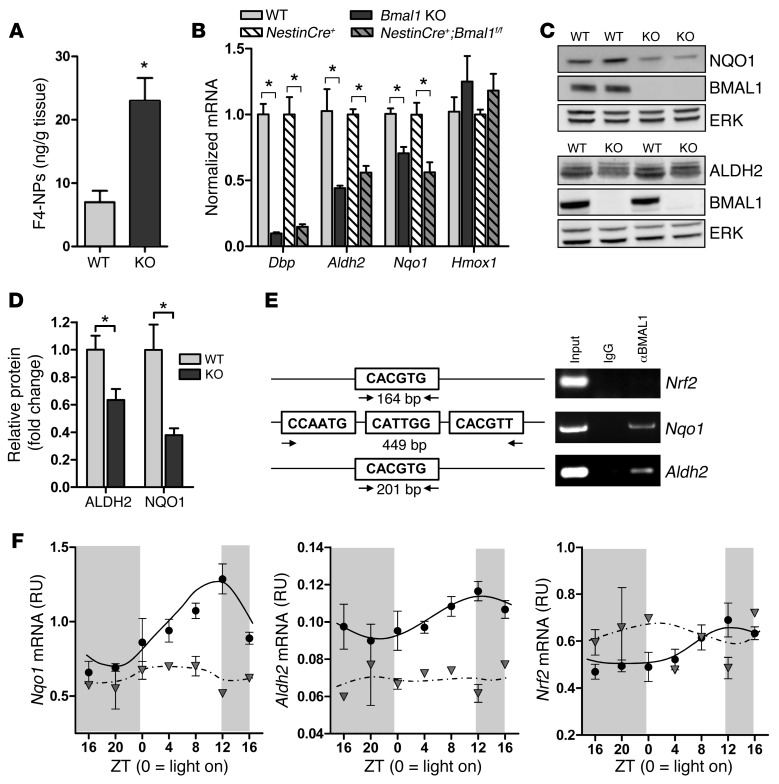
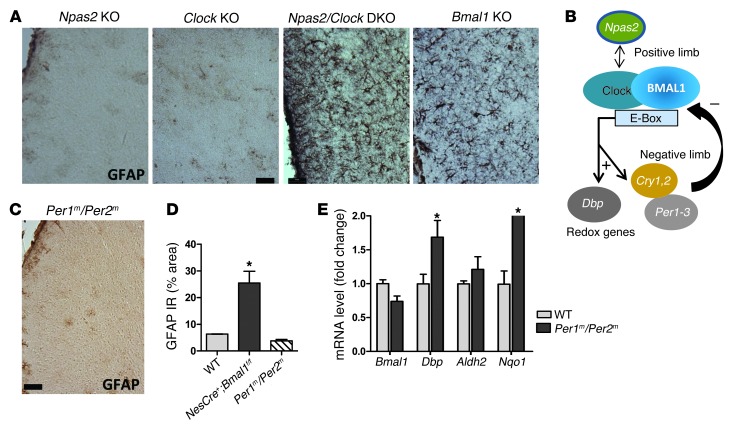
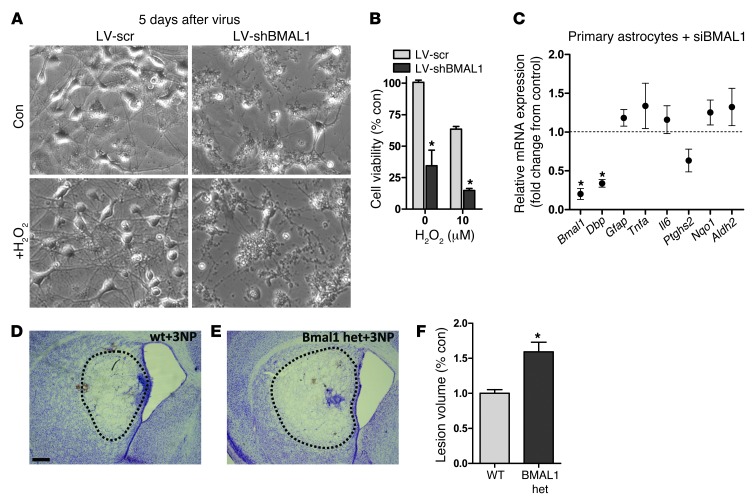
Brain aging is associated with diminished circadian clock output and decreased expression of the core clock proteins, which regulate many aspects of cellular biochemistry and metabolism. The genes encoding clock proteins are expressed throughout the brain, though it is unknown whether these proteins modulate brain homeostasis. We observed that deletion of circadian clock transcriptional activators aryl hydrocarbon receptor nuclear translocator-like (Bmal1) alone, or circadian locomotor output cycles kaput (Clock) in combination with neuronal PAS domain protein 2 (Npas2), induced severe age-dependent astrogliosis in the cortex and hippocampus. Mice lacking the clock gene repressors period circadian clock 1 (Per1) and period circadian clock 2 (Per2) had no observed astrogliosis. Bmal1 deletion caused the degeneration of synaptic terminals and impaired cortical functional connectivity, as well as neuronal oxidative damage and impaired expression of several redox defense genes. Targeted deletion of Bmal1 in neurons and glia caused similar neuropathology, despite the retention of intact circadian behavioral and sleep-wake rhythms. Reduction of Bmal1 expression promoted neuronal death in primary cultures and in mice treated with a chemical inducer of oxidative injury and striatal neurodegeneration. Our findings indicate that BMAL1 in a complex with CLOCK or NPAS2 regulates cerebral redox homeostasis and connects impaired clock gene function to neurodegeneration.
Figures
Comment in
-
Mind your rhythms: an important role for circadian genes in neuroprotection.J Clin Invest. 2013 Dec;123(12):4994-6. doi: 10.1172/JCI73059. Epub 2013 Nov 25. J Clin Invest. 2013. PMID: 24270412 Free PMC article.
References
-
- Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001. 63: 647 676 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 NS057105/NS/NINDS NIH HHS/United States
- R01 NS056125/NS/NINDS NIH HHS/United States
- P01NS074969/NS/NINDS NIH HHS/United States
- P01 NS074969/NS/NINDS NIH HHS/United States
- P50 AG005681/AG/NIA NIH HHS/United States
- NS056125/NS/NINDS NIH HHS/United States
- P30NS057105/NS/NINDS NIH HHS/United States
- HL097800/HL/NHLBI NIH HHS/United States
- R01 HL097800/HL/NHLBI NIH HHS/United States
- R01 NS078223/NS/NINDS NIH HHS/United States
- R25 NS065745/NS/NINDS NIH HHS/United States
- R25NS065745/NS/NINDS NIH HHS/United States
- K08NS079405/NS/NINDS NIH HHS/United States
- K08 NS079405/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
