

High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy
- PMID: 24270810
- PMCID: PMC5841086
- DOI: 10.1038/nature12748
High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy
Abstract
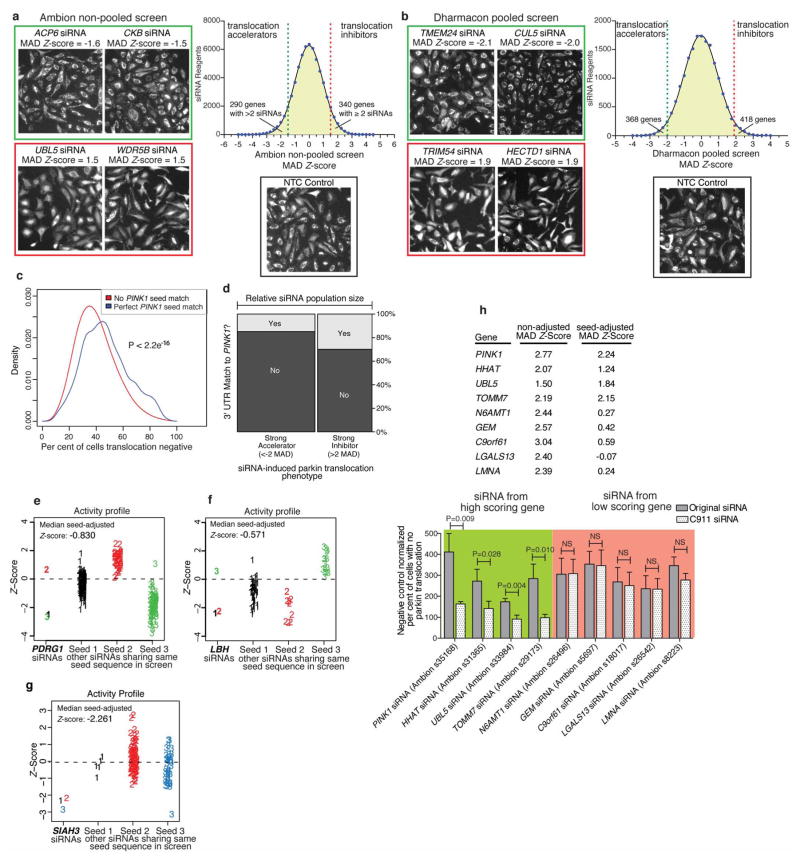
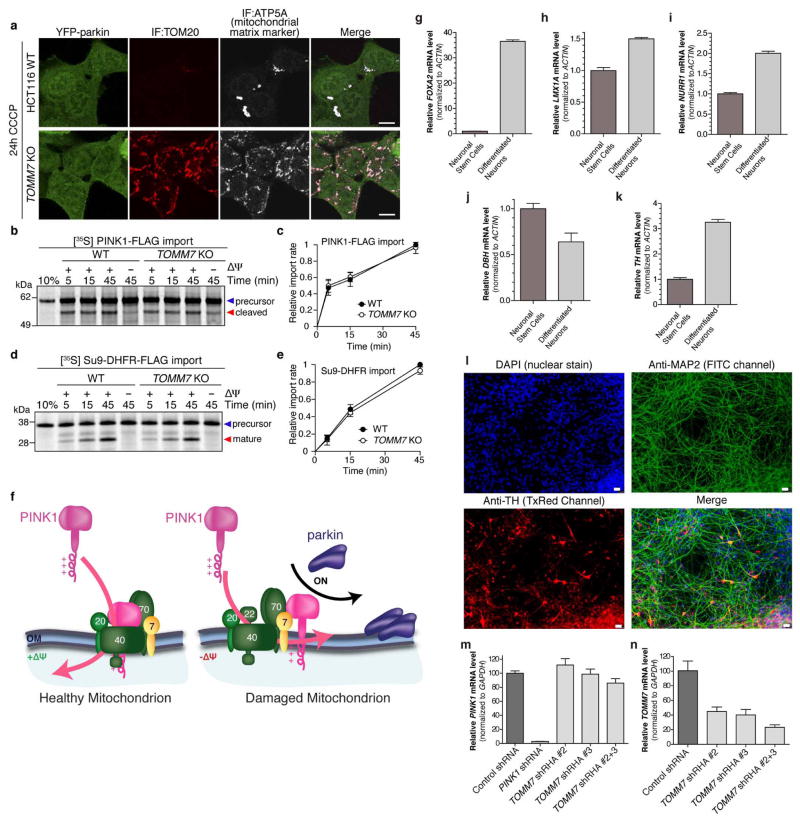
An increasing body of evidence points to mitochondrial dysfunction as a contributor to the molecular pathogenesis of neurodegenerative diseases such as Parkinson's disease. Recent studies of the Parkinson's disease associated genes PINK1 (ref. 2) and parkin (PARK2, ref. 3) indicate that they may act in a quality control pathway preventing the accumulation of dysfunctional mitochondria. Here we elucidate regulators that have an impact on parkin translocation to damaged mitochondria with genome-wide small interfering RNA (siRNA) screens coupled to high-content microscopy. Screening yielded gene candidates involved in diverse cellular processes that were subsequently validated in low-throughput assays. This led to characterization of TOMM7 as essential for stabilizing PINK1 on the outer mitochondrial membrane following mitochondrial damage. We also discovered that HSPA1L (HSP70 family member) and BAG4 have mutually opposing roles in the regulation of parkin translocation. The screens revealed that SIAH3, found to localize to mitochondria, inhibits PINK1 accumulation after mitochondrial insult, reducing parkin translocation. Overall, our screens provide a rich resource to understand mitochondrial quality control.
Conflict of interest statement
The authors declare no competing financial interests.
Figures












References
-
- Schapira AH, Tolosa E. Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nature Rev Neurol. 2010;6:309–317. - PubMed
-
- Valente EM, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. - PubMed
-
- Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
