

Identification of the GPR55 antagonist binding site using a novel set of high-potency GPR55 selective ligands
- PMID: 24274581
- PMCID: PMC3970910
- DOI: 10.1021/bi4008885
Identification of the GPR55 antagonist binding site using a novel set of high-potency GPR55 selective ligands
Abstract
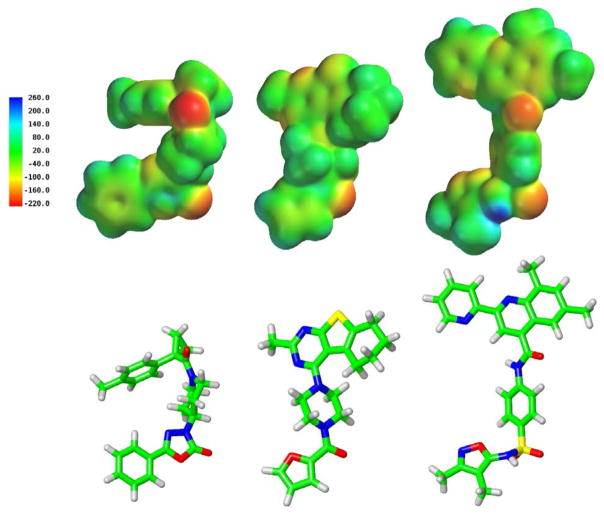
GPR55 is a class A G protein-coupled receptor (GPCR) that has been implicated in inflammatory pain, neuropathic pain, metabolic disorder, bone development, and cancer. Initially deorphanized as a cannabinoid receptor, GPR55 has been shown to be activated by non-cannabinoid ligands such as l-α-lysophosphatidylinositol (LPI). While there is a growing body of evidence of physiological and pathophysiological roles for GPR55, the paucity of specific antagonists has limited its study. In collaboration with the Molecular Libraries Probe Production Centers Network initiative, we identified a series of GPR55 antagonists using a β-arrestin, high-throughput, high-content screen of ~300000 compounds. This screen yielded novel, GPR55 antagonist chemotypes with IC50 values in the range of 0.16-2.72 μM [Heynen-Genel, S., et al. (2010) Screening for Selective Ligands for GPR55: Antagonists (ML191, ML192, ML193) (Bookshelf ID NBK66153; PMID entry 22091481)]. Importantly, many of the GPR55 antagonists were completely selective, with no agonism or antagonism against GPR35, CB1, or CB2 up to 20 μM. Using a model of the GPR55 inactive state, we studied the binding of an antagonist series that emerged from this screen. These studies suggest that GPR55 antagonists possess a head region that occupies a horizontal binding pocket extending into the extracellular loop region, a central ligand portion that fits vertically in the receptor binding pocket and terminates with a pendant aromatic or heterocyclic ring that juts out. Both the region that extends extracellularly and the pendant ring are features associated with antagonism. Taken together, our results provide a set of design rules for the development of second-generation GPR55 selective antagonists.
Figures









References
-
- Staton PC, Hatcher JP, Walker DJ, Morrison AD, Shapland EM, Hughes JP, Chong E, Mander PK, Green PJ, Billinton A, Fulleylove M, Lancaster HC, Smith JC, Bailey LT, Wise A, Brown AJ, Richardson JC, Chessell IP. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. 2008;139:225–236. - PubMed
-
- Andradas C, Caffarel MM, Perez-Gomez E, Salazar M, Lorente M, Velasco G, Guzman M, Sanchez C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene. 2011;30:245–252. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54 HG005033/HG/NHGRI NIH HHS/United States
- U01 DA022950/DA/NIDA NIH HHS/United States
- DA023204/DA/NIDA NIH HHS/United States
- P50 DA005274/DA/NIDA NIH HHS/United States
- P30 DA013429/DA/NIDA NIH HHS/United States
- R21 DA029432/DA/NIDA NIH HHS/United States
- DA029432/DA/NIDA NIH HHS/United States
- X01 DA026205/DA/NIDA NIH HHS/United States
- DA021358/DA/NIDA NIH HHS/United States
- R01 DA023204/DA/NIDA NIH HHS/United States
- U54HG005033/HG/NHGRI NIH HHS/United States
- R21 NS077347/NS/NINDS NIH HHS/United States
- NS077347/NS/NINDS NIH HHS/United States
- DA022950/DA/NIDA NIH HHS/United States
- K05 DA021358/DA/NIDA NIH HHS/United States
- P30DA013429/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
