

Spontaneous generation of infectious prion disease in transgenic mice
- PMID: 24274622
- PMCID: PMC3840888
- DOI: 10.3201/eid1912.130106
Spontaneous generation of infectious prion disease in transgenic mice
Abstract
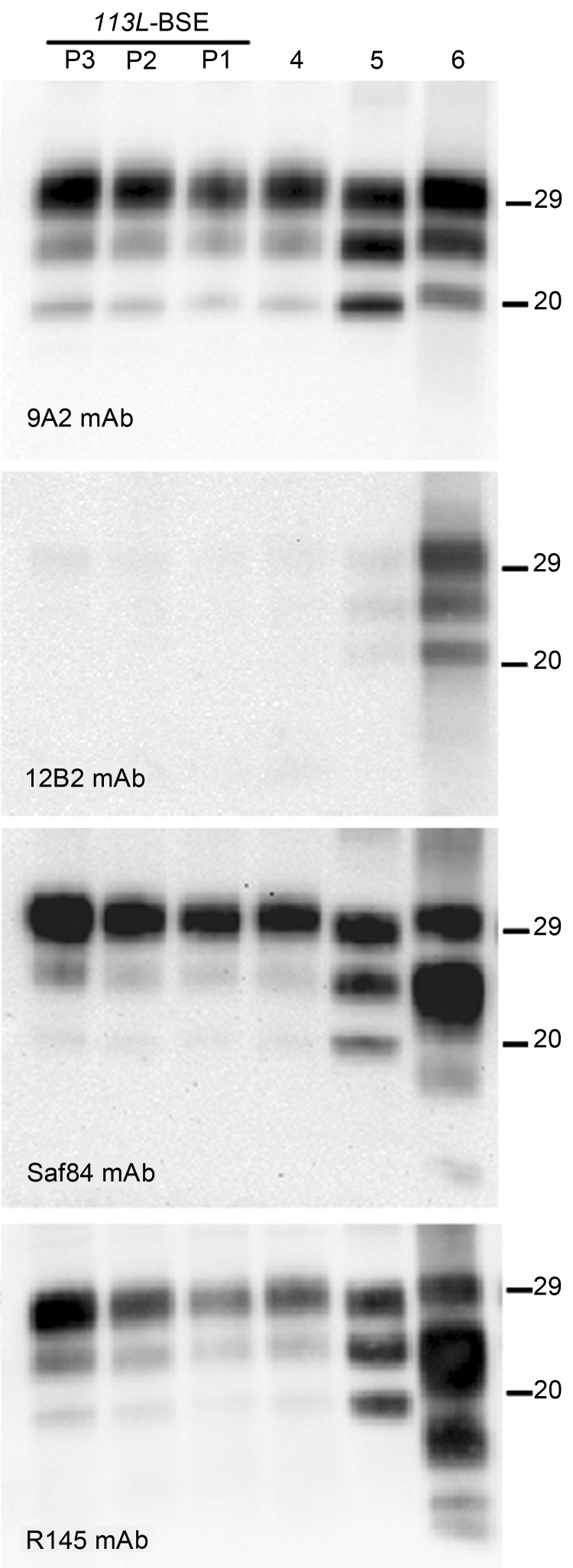
We generated transgenic mice expressing bovine cellular prion protein (PrP(C)) with a leucine substitution at codon 113 (113L). This protein is homologous to human protein with mutation 102L, and its genetic link with Gerstmann-Sträussler-Scheinker syndrome has been established. This mutation in bovine PrP(C) causes a fully penetrant, lethal, spongiform encephalopathy. This genetic disease was transmitted by intracerebral inoculation of brain homogenate from ill mice expressing mutant bovine PrP to mice expressing wild-type bovine PrP, which indicated de novo generation of infectious prions. Our findings demonstrate that a single amino acid change in the PrP(C) sequence can induce spontaneous generation of an infectious prion disease that differs from all others identified in hosts expressing the same PrP(C) sequence. These observations support the view that a variety of infectious prion strains might spontaneously emerge in hosts displaying random genetic PrP(C) mutations.
Keywords: BSE; Gerstmann–Sträussler–Scheinker syndrome; TSE; bovine PrP; bovine prion protein; bovine spongiform encephalopathy; prions and related diseases; spontaneous generation; transgenic mice; transmissible spongiform encephalopathy.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials