

A direct interaction between leucine-rich repeat kinase 2 and specific β-tubulin isoforms regulates tubulin acetylation
- PMID: 24275654
- PMCID: PMC3887213
- DOI: 10.1074/jbc.M113.507913
A direct interaction between leucine-rich repeat kinase 2 and specific β-tubulin isoforms regulates tubulin acetylation
Abstract
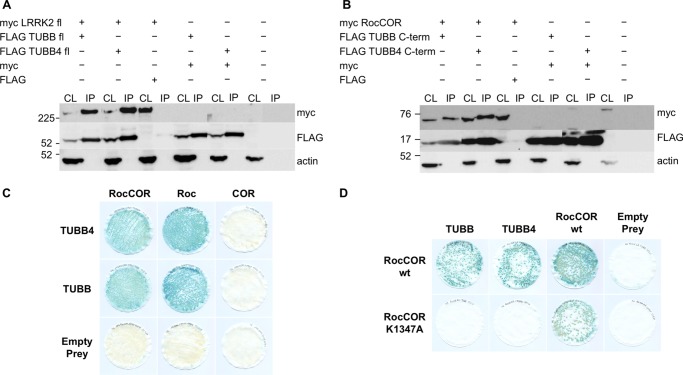
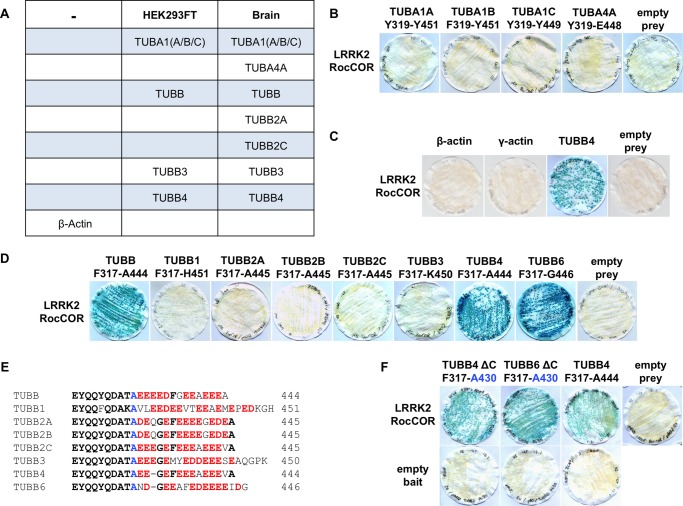
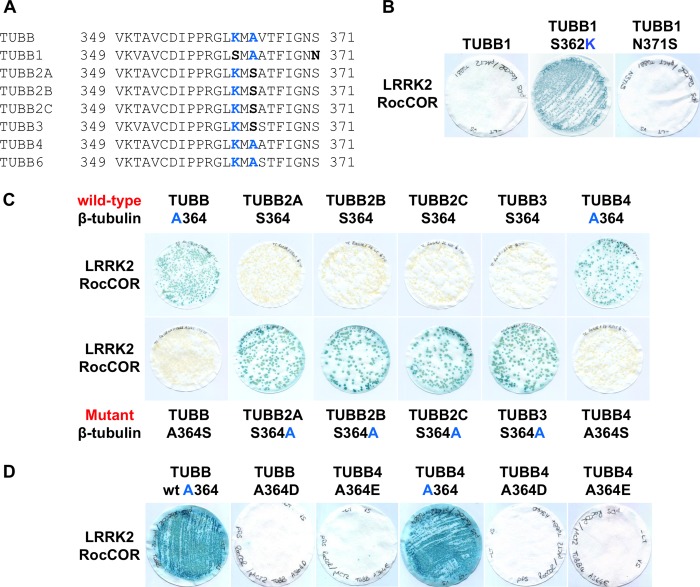
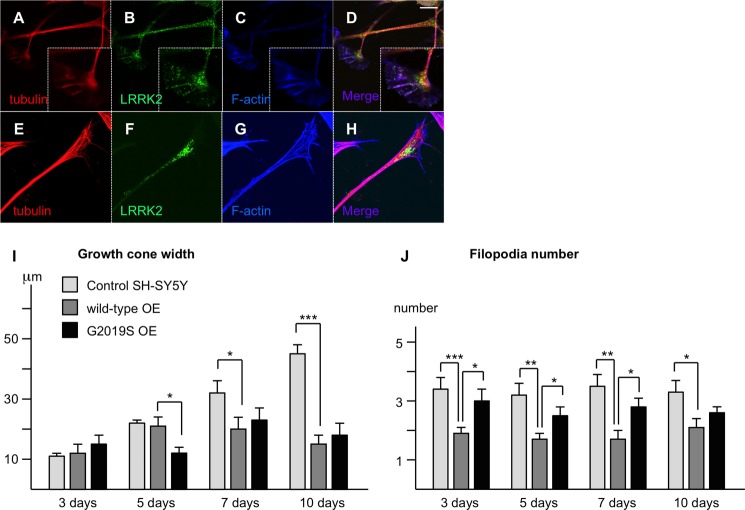
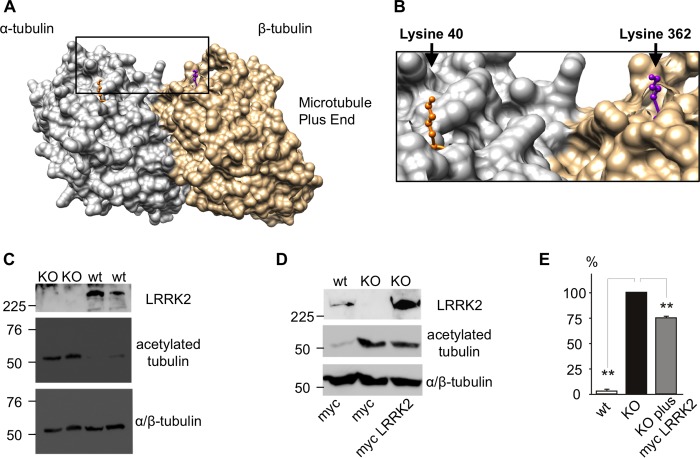
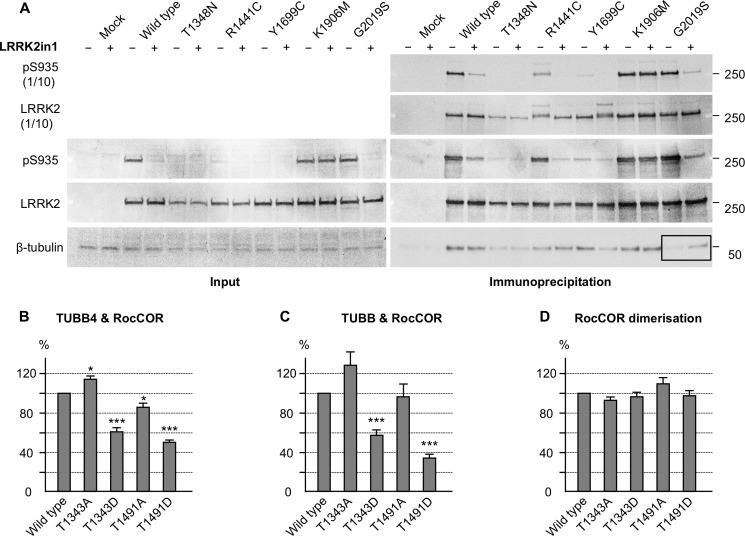
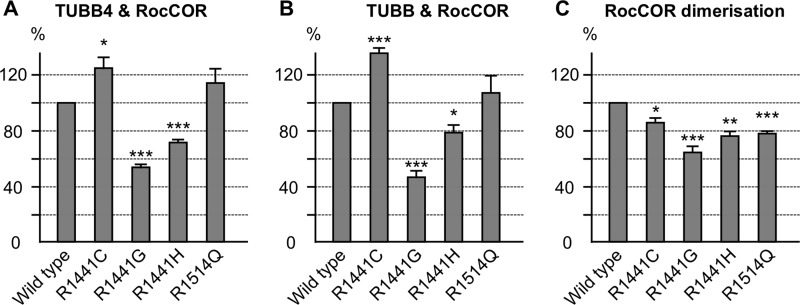
Mutations in LRRK2, encoding the multifunctional protein leucine-rich repeat kinase 2 (LRRK2), are a common cause of Parkinson disease. LRRK2 has been suggested to influence the cytoskeleton as LRRK2 mutants reduce neurite outgrowth and cause an accumulation of hyperphosphorylated Tau. This might cause alterations in the dynamic instability of microtubules suggested to contribute to the pathogenesis of Parkinson disease. Here, we describe a direct interaction between LRRK2 and β-tubulin. This interaction is conferred by the LRRK2 Roc domain and is disrupted by the familial R1441G mutation and artificial Roc domain mutations that mimic autophosphorylation. LRRK2 selectively interacts with three β-tubulin isoforms: TUBB, TUBB4, and TUBB6, one of which (TUBB4) is mutated in the movement disorder dystonia type 4 (DYT4). Binding specificity is determined by lysine 362 and alanine 364 of β-tubulin. Molecular modeling was used to map the interaction surface to the luminal face of microtubule protofibrils in close proximity to the lysine 40 acetylation site in α-tubulin. This location is predicted to be poorly accessible within mature stabilized microtubules, but exposed in dynamic microtubule populations. Consistent with this finding, endogenous LRRK2 displays a preferential localization to dynamic microtubules within growth cones, rather than adjacent axonal microtubule bundles. This interaction is functionally relevant to microtubule dynamics, as mouse embryonic fibroblasts derived from LRRK2 knock-out mice display increased microtubule acetylation. Taken together, our data shed light on the nature of the LRRK2-tubulin interaction, and indicate that alterations in microtubule stability caused by changes in LRRK2 might contribute to the pathogenesis of Parkinson disease.
Keywords: Cytoskeletal Dynamics; GTPase Mutation; Growth Cone; Lrrk2; Microtubules; Molecular Genetics; Parkinson Disease; RocCOR; Tubulin; Tubulin Acetylation.
Figures


References
-
- Paisán-Ruíz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., Johnson J., Martinez J. R., Nicholl D., Carrera I. M., Pena A. S., de Silva R., Lees A., Martí-Massó J. F., Pérez-Tur J., Wood N. W., Singleton A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600 - PubMed
-
- Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., Asmus F., Müller-Myhsok B., Dickson D. W., Meitinger T., Strom T. M., Wszolek Z. K., Gasser T. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 - PubMed
-
- Ross O. A., Toft M., Whittle A. J., Johnson J. L., Papapetropoulos S., Mash D. C., Litvan I., Gordon M. F., Wszolek Z. K., Farrer M. J., Dickson D. W. (2006) Lrrk2 and Lewy body disease. Ann. Neurol. 59, 388–393 - PubMed
-
- Kumari U., Tan E. K. (2009) LRRK2 in Parkinson's disease. Genetic and clinical studies from patients. FEBS J. 276, 6455–6463 - PubMed
-
- Berwick D. C., Harvey K. (2011) LRRK2 signaling pathways. The key to unlocking neurodegeneration? Trends Cell Biol. 21, 257–265 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
