

S6K1 negatively regulates TAK1 activity in the toll-like receptor signaling pathway
- PMID: 24277938
- PMCID: PMC3911500
- DOI: 10.1128/MCB.01225-13
S6K1 negatively regulates TAK1 activity in the toll-like receptor signaling pathway
Abstract
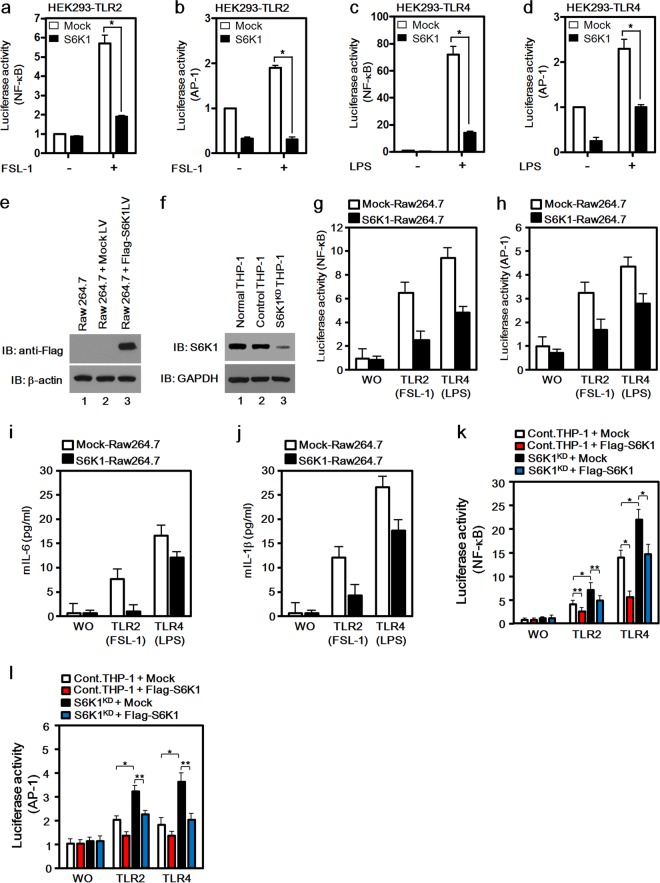
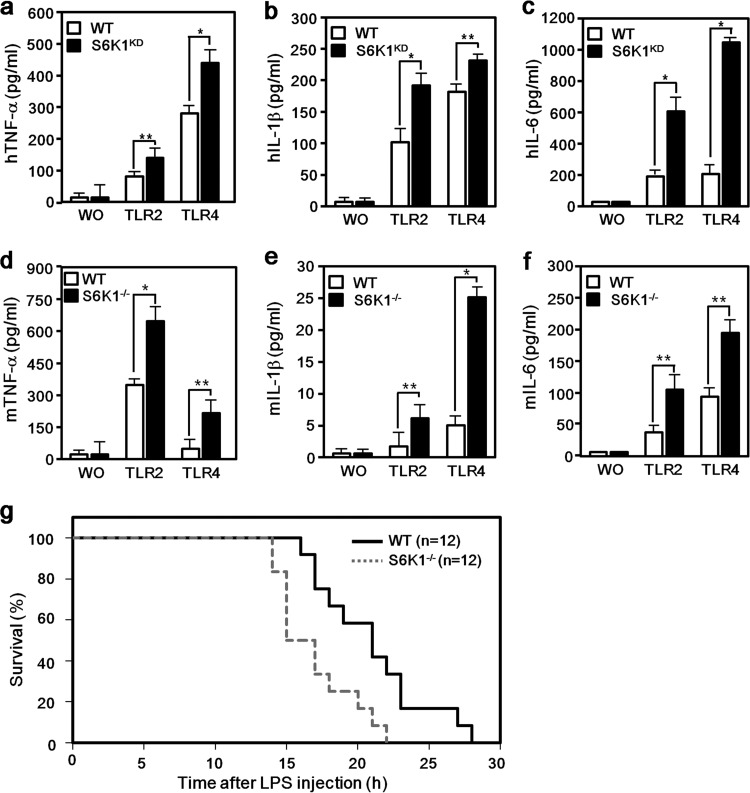
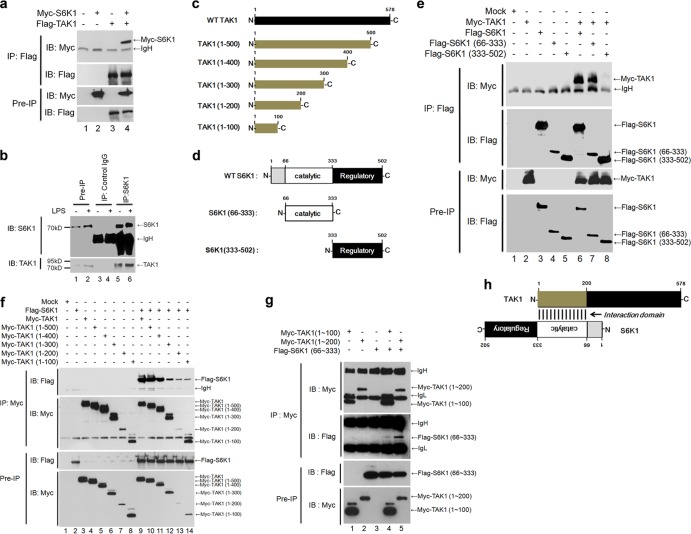
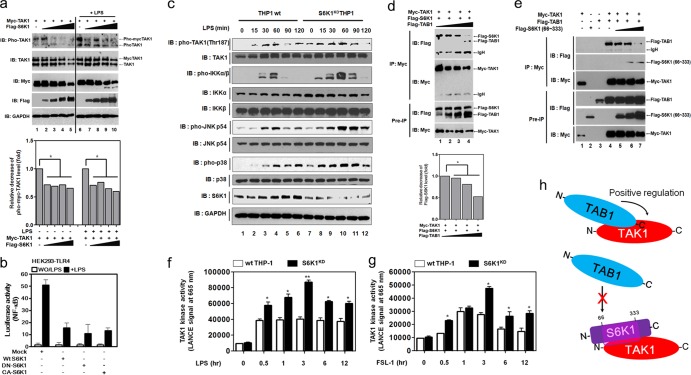
Transforming growth factor β (TGF-β)-activated kinase 1 (TAK1) is a key regulator in the signals transduced by proinflammatory cytokines and Toll-like receptors (TLRs). The regulatory mechanism of TAK1 in response to various tissue types and stimuli remains incompletely understood. Here, we show that ribosomal S6 kinase 1 (S6K1) negatively regulates TLR-mediated signals by inhibiting TAK1 activity. S6K1 overexpression causes a marked reduction in NF-κB and AP-1 activity induced by stimulation of TLR2 or TLR4. In contrast, S6K1(-/-) and S6K1 knockdown cells display enhanced production of inflammatory cytokines. Moreover, S6K1(-/-) mice exhibit decreased survival in response to challenge with lipopolysaccharide (LPS). We found that S6K1 inhibits TAK1 kinase activity by interfering with the interaction between TAK1 and TAB1, which is a key regulator protein for TAK1 catalytic function. Upon stimulation with TLR ligands, S6K1 deficiency causes a marked increase in TAK1 kinase activity that in turn induces a substantial enhancement of NF-κB-dependent gene expression, indicating that S6K1 is negatively involved in the TLR signaling pathway by the inhibition of TAK1 activity. Our findings contribute to understanding the molecular pathogenesis of the impaired immune responses seen in type 2 diabetes, where S6K1 plays a key role both in driving insulin resistance and modulating TLR signaling.
Figures
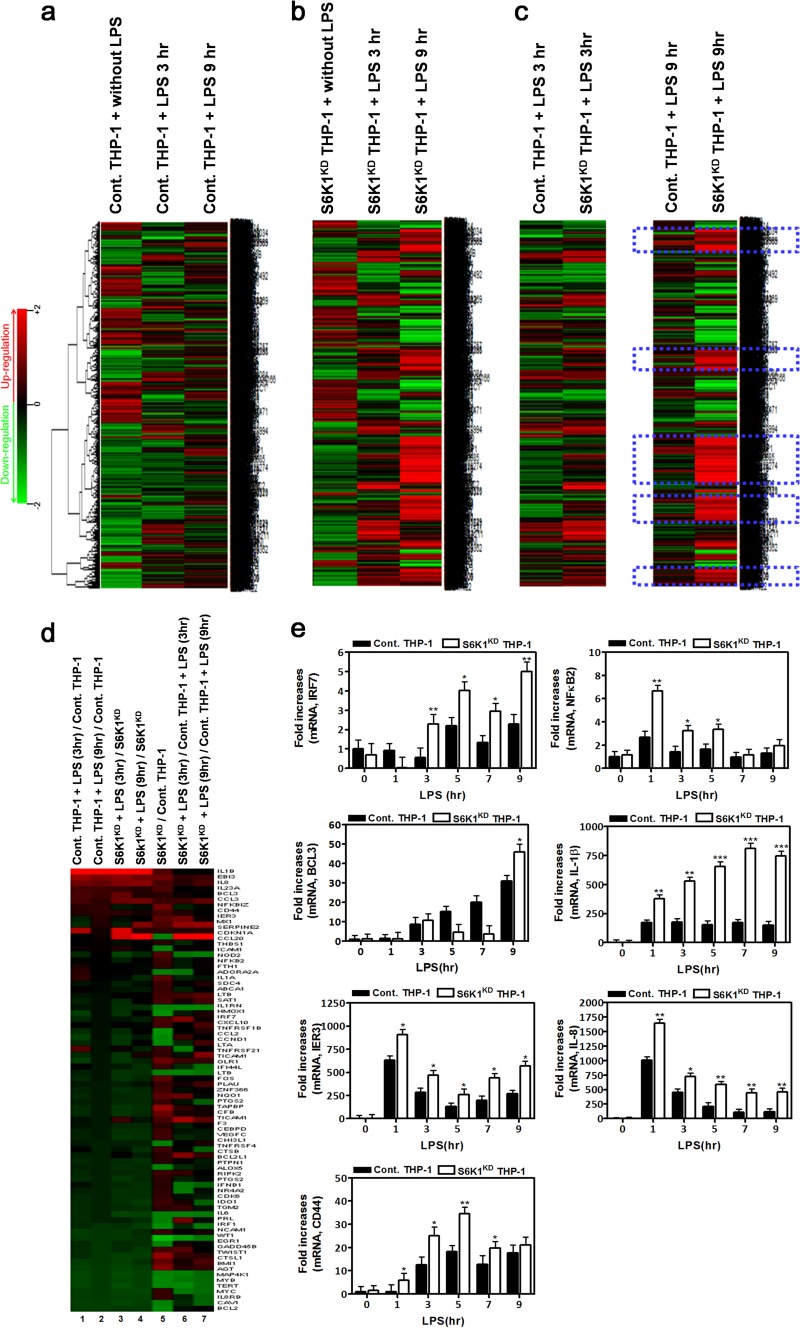
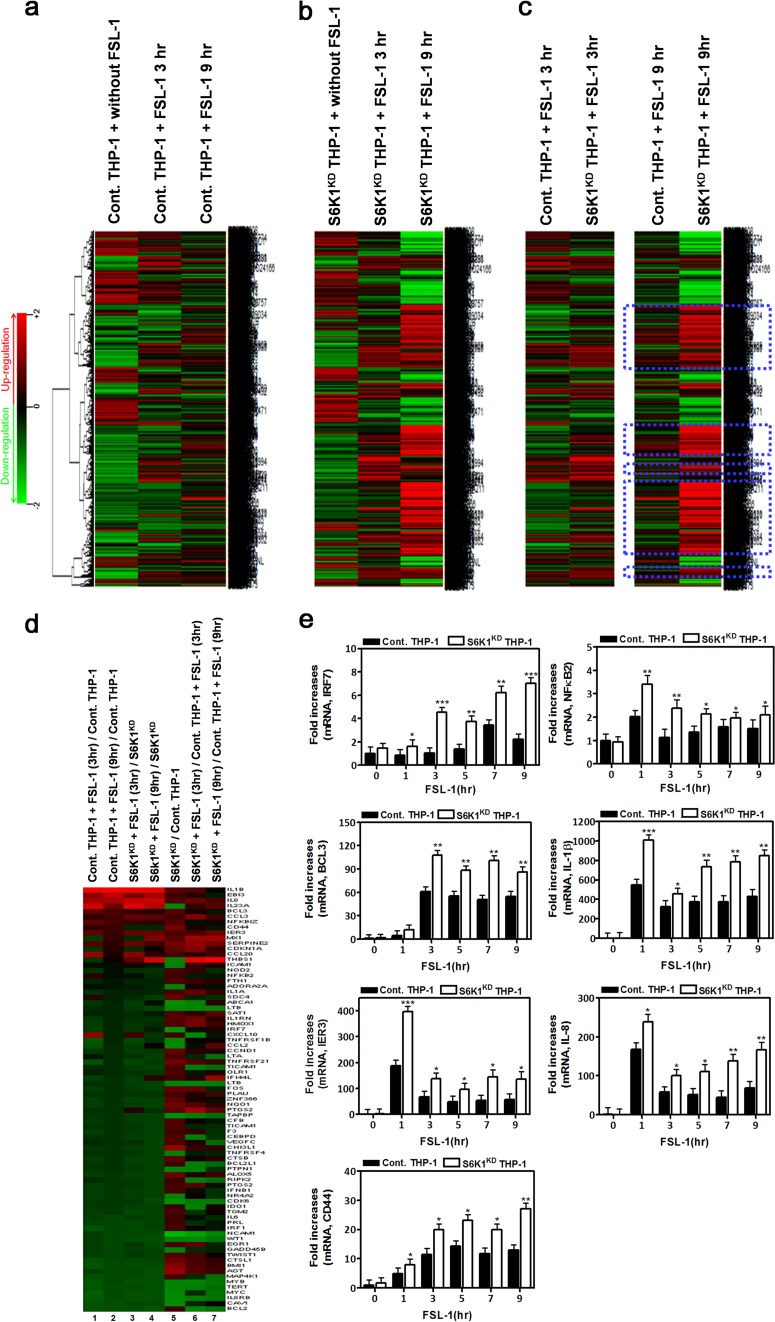
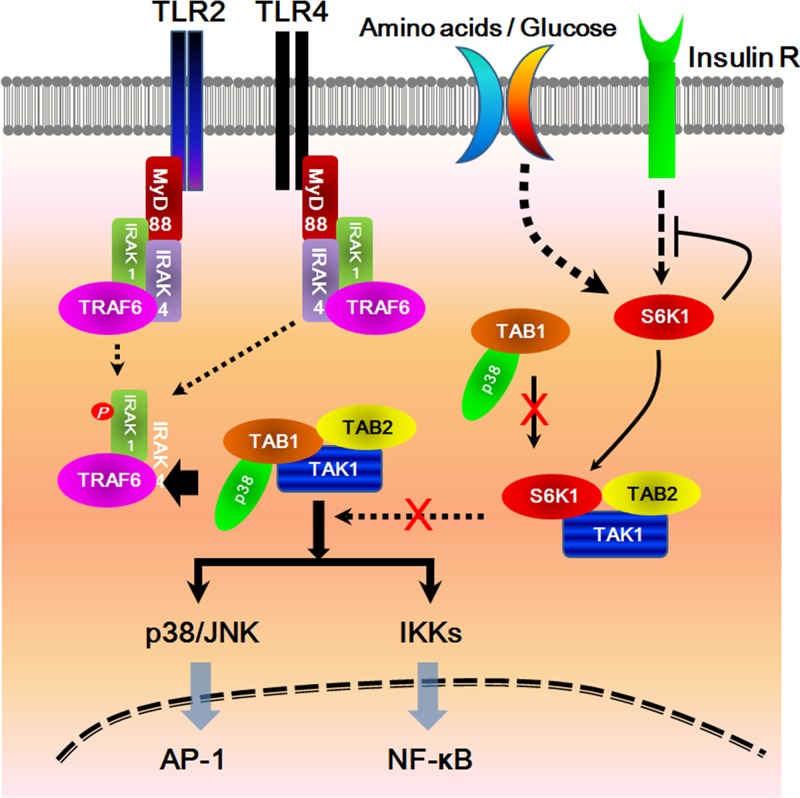
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous