

Systematic analysis of compositional order of proteins reveals new characteristics of biological functions and a universal correlate of macroevolution
- PMID: 24278003
- PMCID: PMC3836704
- DOI: 10.1371/journal.pcbi.1003346
Systematic analysis of compositional order of proteins reveals new characteristics of biological functions and a universal correlate of macroevolution
Abstract
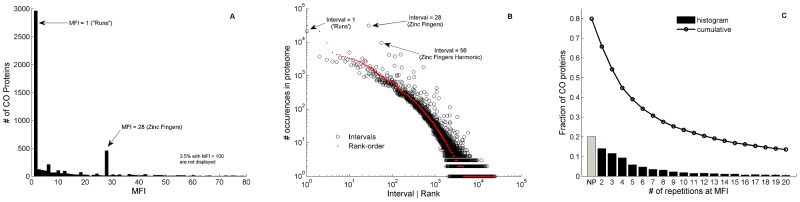
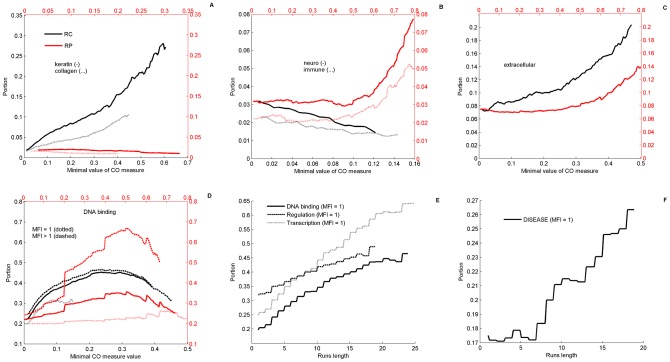
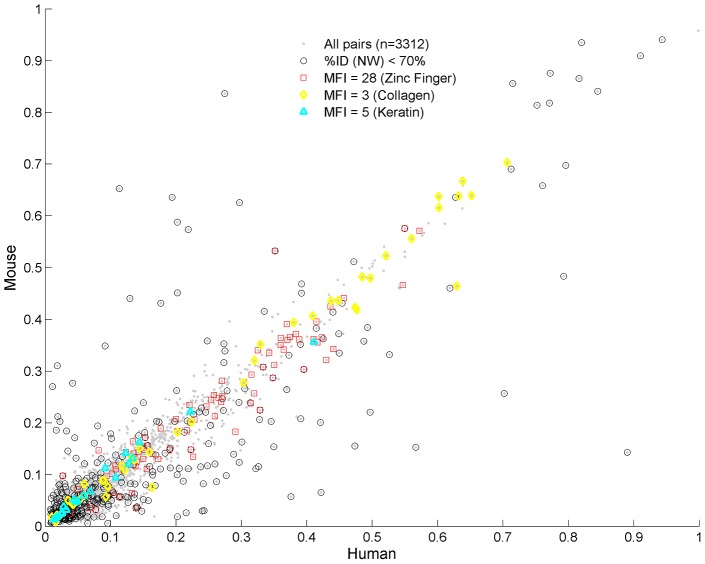
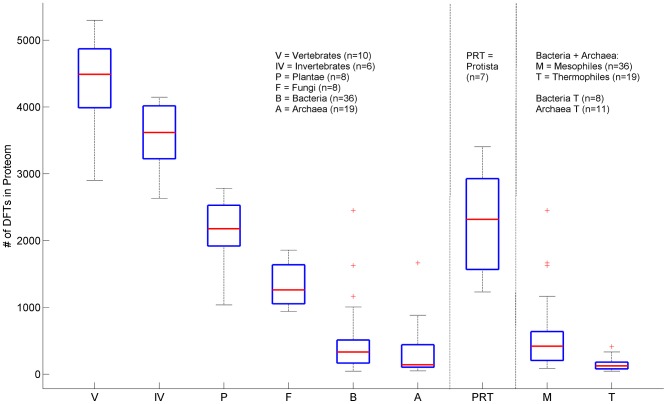
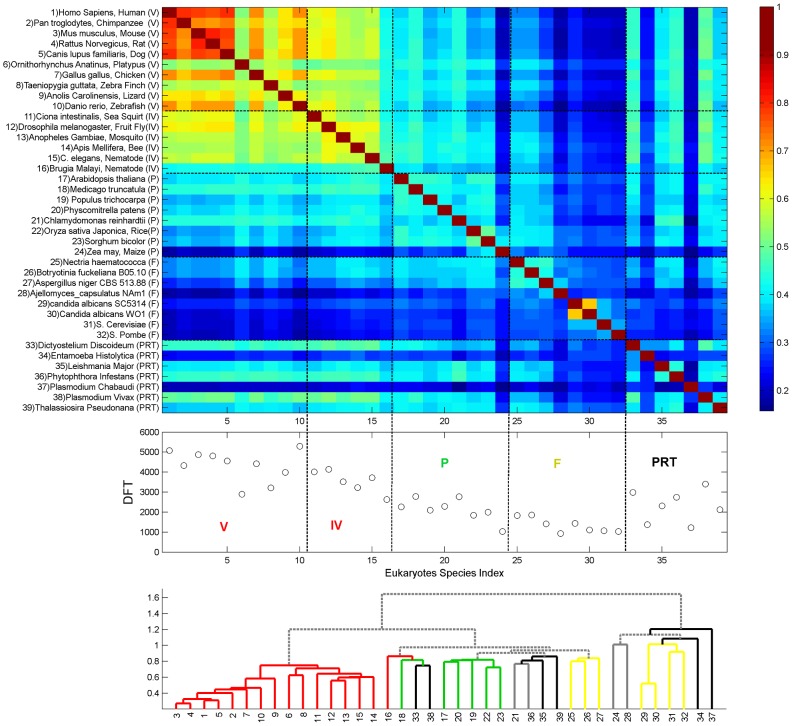
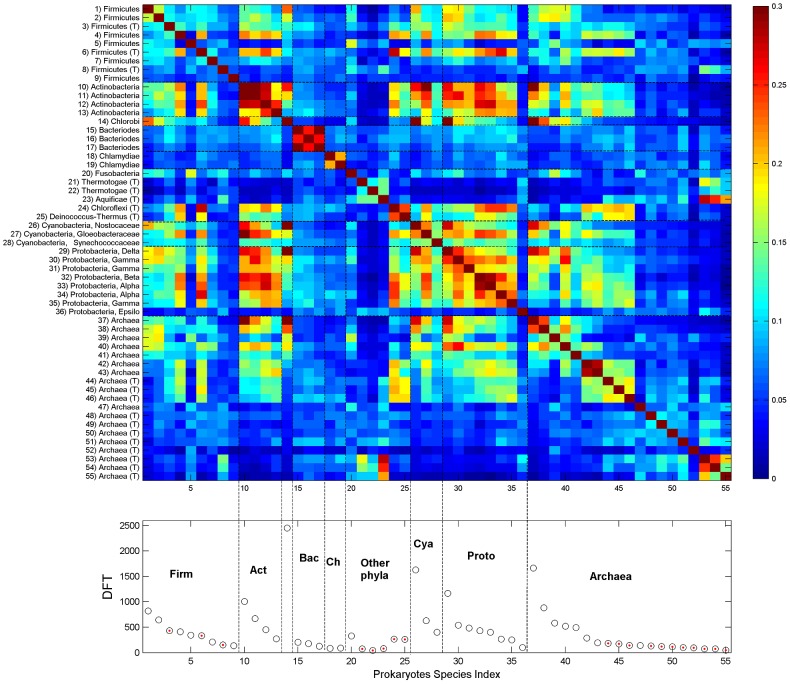
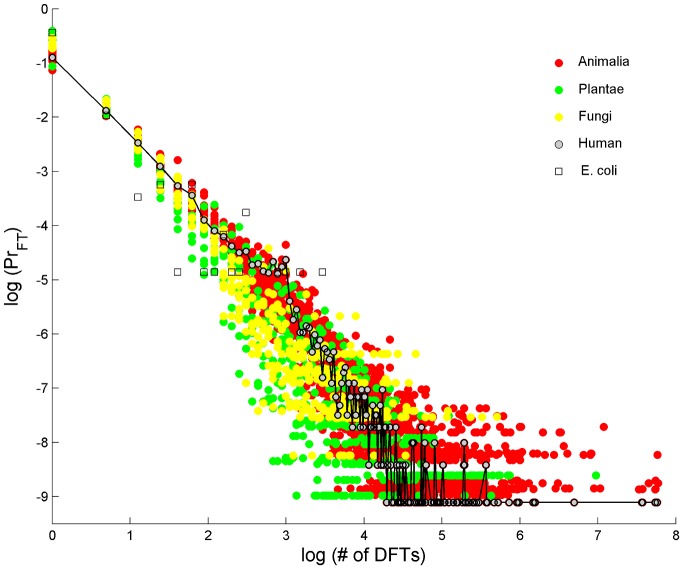
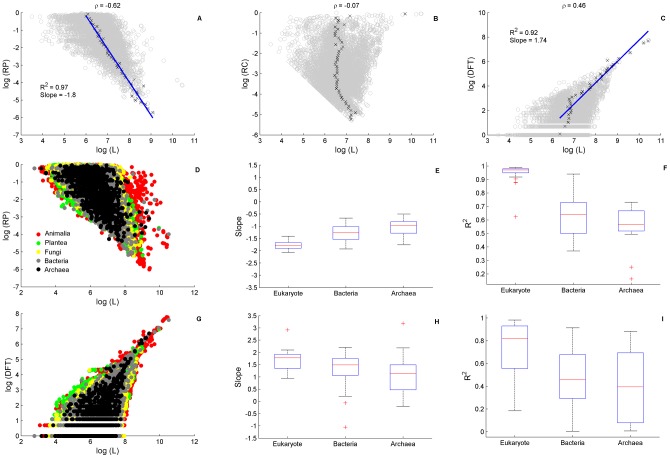
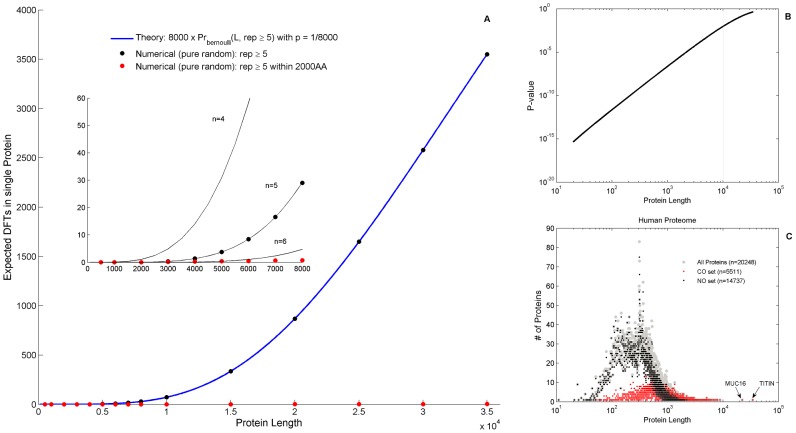
We present a novel analysis of compositional order (CO) based on the occurrence of Frequent amino-acid Triplets (FTs) that appear much more than random in protein sequences. The method captures all types of proteomic compositional order including single amino-acid runs, tandem repeats, periodic structure of motifs and otherwise low complexity amino-acid regions. We introduce new order measures, distinguishing between 'regularity', 'periodicity' and 'vocabulary', to quantify these phenomena and to facilitate the identification of evolutionary effects. Detailed analysis of representative species across the tree-of-life demonstrates that CO proteins exhibit numerous functional enrichments, including a wide repertoire of particular patterns of dependencies on regularity and periodicity. Comparison between human and mouse proteomes further reveals the interplay of CO with evolutionary trends, such as faster substitution rate in mouse leading to decrease of periodicity, while innovation along the human lineage leads to larger regularity. Large-scale analysis of 94 proteomes leads to systematic ordering of all major taxonomic groups according to FT-vocabulary size. This is measured by the count of Different Frequent Triplets (DFT) in proteomes. The latter provides a clear hierarchical delineation of vertebrates, invertebrates, plants, fungi and prokaryotes, with thermophiles showing the lowest level of FT-vocabulary. Among eukaryotes, this ordering correlates with phylogenetic proximity. Interestingly, in all kingdoms CO accumulation in the proteome has universal characteristics. We suggest that CO is a genomic-information correlate of both macroevolution and various protein functions. The results indicate a mechanism of genomic 'innovation' at the peptide level, involved in protein elongation, shaped in a universal manner by mutational and selective forces.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Context characterization of amino acid homorepeats using evolution, position, and order.Proteins. 2017 Apr;85(4):709-719. doi: 10.1002/prot.25250. Epub 2017 Feb 6. Proteins. 2017. PMID: 28097686
-
COPASAAR--a database for proteomic analysis of single amino acid repeats.BMC Bioinformatics. 2005 Aug 3;6:196. doi: 10.1186/1471-2105-6-196. BMC Bioinformatics. 2005. PMID: 16078990 Free PMC article.
-
C-terminal motif prediction in eukaryotic proteomes using comparative genomics and statistical over-representation across protein families.BMC Genomics. 2007 Jun 26;8:191. doi: 10.1186/1471-2164-8-191. BMC Genomics. 2007. PMID: 17594486 Free PMC article.
-
Protein family classification and functional annotation.Comput Biol Chem. 2003 Feb;27(1):37-47. doi: 10.1016/s1476-9271(02)00098-1. Comput Biol Chem. 2003. PMID: 12798038 Review.
-
The use of evolutionary patterns in protein annotation.Curr Opin Struct Biol. 2012 Jun;22(3):316-25. doi: 10.1016/j.sbi.2012.05.001. Epub 2012 May 24. Curr Opin Struct Biol. 2012. PMID: 22633559 Free PMC article. Review.
Cited by
-
The overdue promise of short tandem repeat variation for heritability.Trends Genet. 2014 Nov;30(11):504-12. doi: 10.1016/j.tig.2014.07.008. Epub 2014 Aug 30. Trends Genet. 2014. PMID: 25182195 Free PMC article. Review.
-
Mutation-selection balance and compensatory mechanisms in tumour evolution.Nat Rev Genet. 2021 Apr;22(4):251-262. doi: 10.1038/s41576-020-00299-4. Epub 2020 Nov 30. Nat Rev Genet. 2021. PMID: 33257848 Review.
-
Positive and strongly relaxed purifying selection drive the evolution of repeats in proteins.Nat Commun. 2016 Nov 18;7:13570. doi: 10.1038/ncomms13570. Nat Commun. 2016. PMID: 27857066 Free PMC article.
-
Lineage-specific protein repeat expansions and contractions reveal malleable regions of immune genes.Genes Immun. 2022 Nov;23(7):218-234. doi: 10.1038/s41435-022-00186-4. Epub 2022 Oct 6. Genes Immun. 2022. PMID: 36203090
-
Compensatory relationship between low-complexity regions and gene paralogy in the evolution of prokaryotes.Proc Natl Acad Sci U S A. 2023 Apr 18;120(16):e2300154120. doi: 10.1073/pnas.2300154120. Epub 2023 Apr 10. Proc Natl Acad Sci U S A. 2023. PMID: 37036997 Free PMC article.
References
-
- Koonin EV, Wolf YI, Karev GP (2002) The structure of the protein universe and genome evolution. Nature 420: 218–223. - PubMed
-
- Wootton JC, Federhen S (1993) Statistics of local complexity in amino acid sequences and sequence databases. Computers Chem 17 (2) 149–163.
-
- Levinson G, Gutman GA (1987) Slipped-Strand Mispairing: A Major Mechanism for DNA Sequence Evolution. Mol Biol Evol 4 (3) 203:221. - PubMed
-
- Wootton JC (1994) Sequences with ‘unusual’ amino acid compositions. Curr Opinion Struct Biology 4: 413–421.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
