

Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia
- PMID: 24278020
- PMCID: PMC3836735
- DOI: 10.1371/journal.ppat.1003773
Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia
Abstract
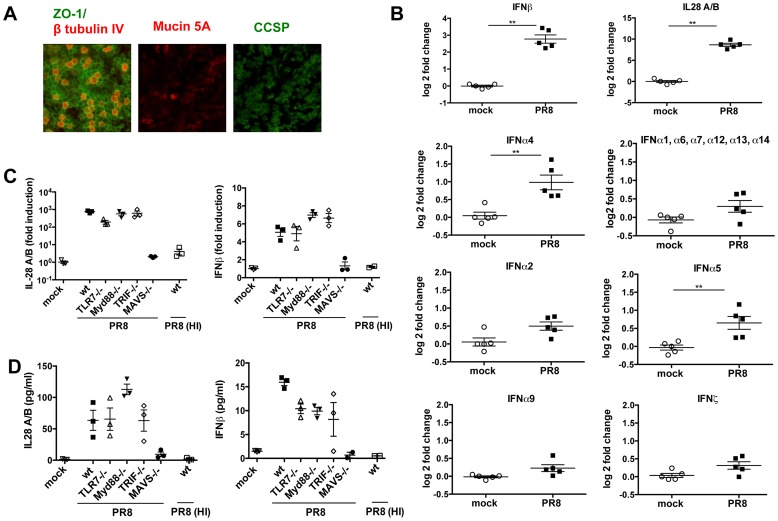
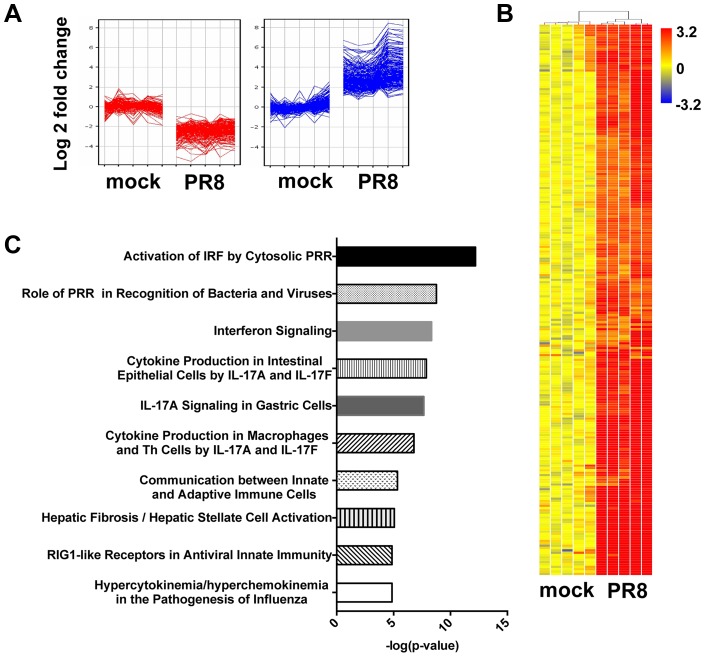
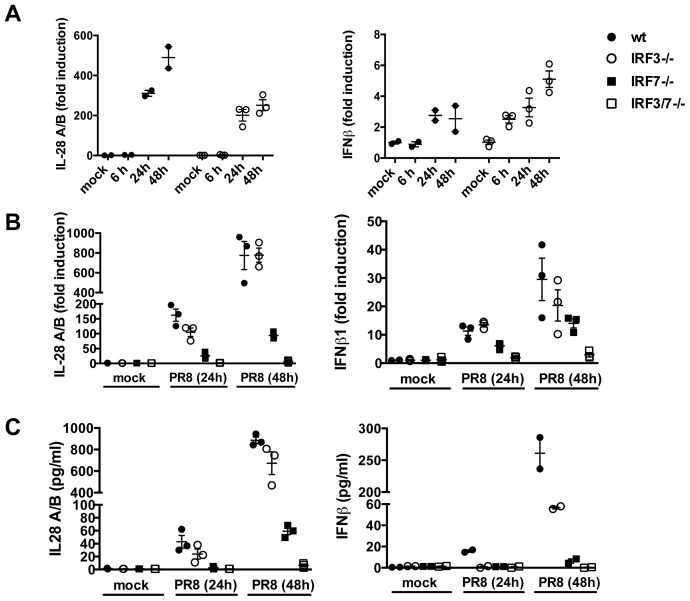
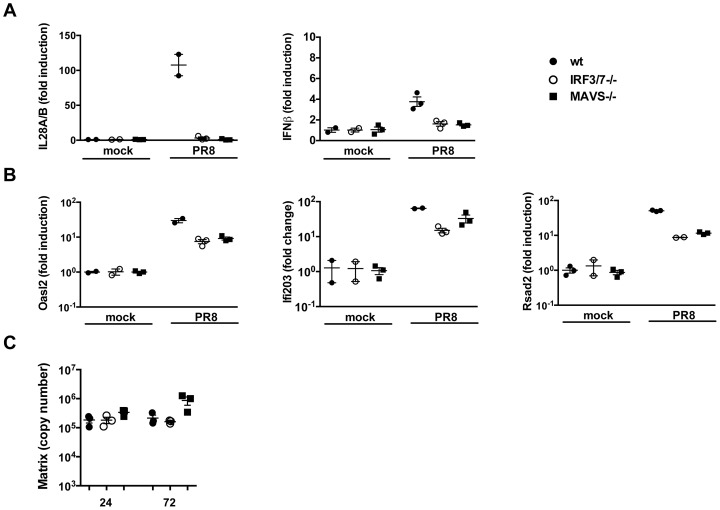
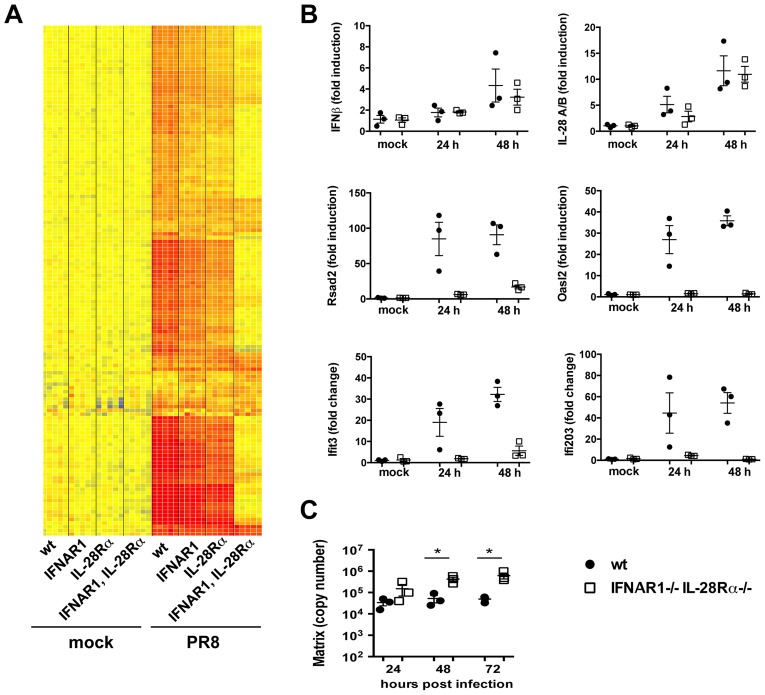
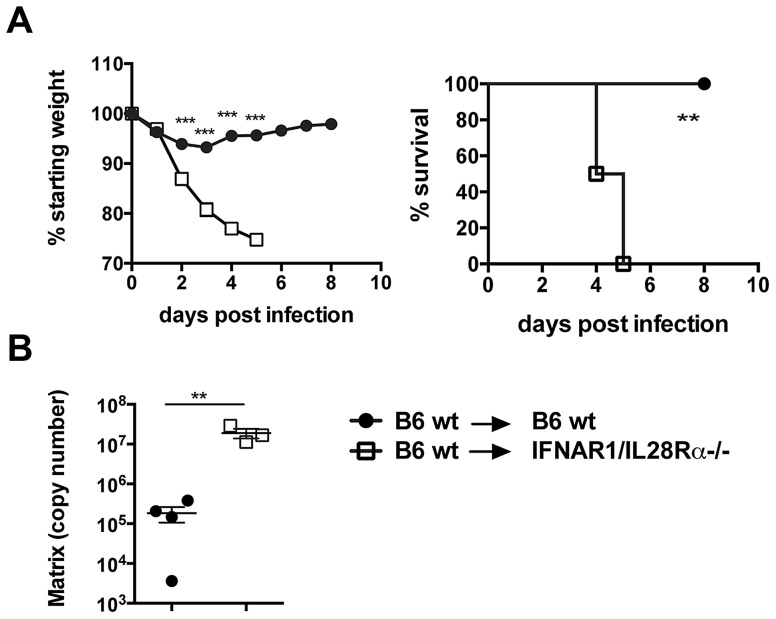
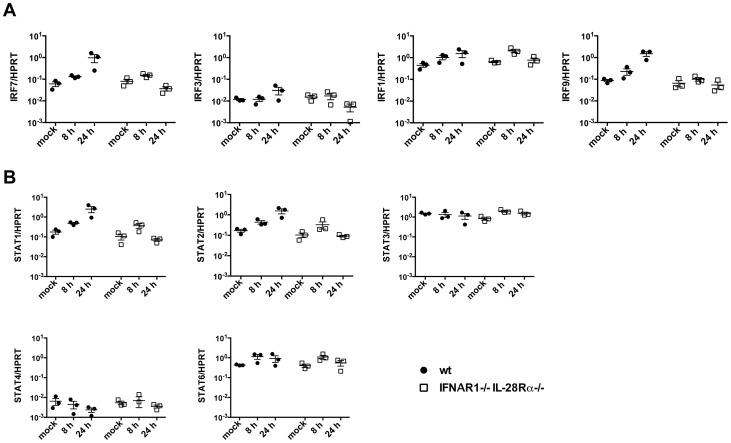
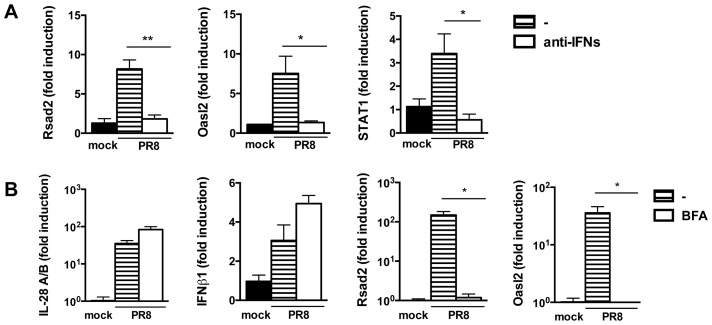
Interferons (IFNs) are a group of cytokines with a well-established antiviral function. They can be induced by viral infection, are secreted and bind to specific receptors on the same or neighbouring cells to activate the expression of hundreds of IFN stimulated genes (ISGs) with antiviral function. Type I IFN has been known for more than half a century. However, more recently, type III IFN (IFNλ, IL-28/29) was shown to play a similar role and to be particularly important at epithelial surfaces. Here we show that airway epithelia, the primary target of influenza A virus, produce both IFN I and III upon infection, and that induction of both depends on the RIG-I/MAVS pathway. While IRF3 is generally regarded as the transcription factor required for initiation of IFN transcription and the so-called "priming loop", we find that IRF3 deficiency has little impact on IFN expression. In contrast, lack of IRF7 reduced IFN production significantly, and only IRF3(-/-)IRF7(-/-) double deficiency completely abolished it. The transcriptional response to influenza infection was largely dependent on IFNs, as it was reduced to a few upregulated genes in epithelia lacking receptors for both type I and III IFN (IFNAR1(-/-)IL-28Rα(-/-)). Wild-type epithelia and epithelia deficient in either the type I IFN receptor or the type III IFN receptor exhibit similar transcriptional profiles in response to virus, indicating that none of the induced genes depends selectively on only one IFN system. In chimeric mice, the lack of both IFN I and III signalling in the stromal compartment alone significantly increased the susceptibility to influenza infection. In conclusion, virus infection of airway epithelia induces, via a RIG-I/MAVS/IRF7 dependent pathway, both type I and III IFNs which drive two completely overlapping and redundant amplification loops to upregulate ISGs and protect from influenza infection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Isaacs A, Lindenmann J (1957) Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147: 258–267. - PubMed
-
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, et al. (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23: 19–28. - PubMed
-
- Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, et al. (2010) RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140: 397–408. - PubMed
-
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, et al. (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6: 981–988. - PubMed
-
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, et al. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
