

Mapping genome-wide transcription factor binding sites in frozen tissues
- PMID: 24279905
- PMCID: PMC3848595
- DOI: 10.1186/1756-8935-6-30
Mapping genome-wide transcription factor binding sites in frozen tissues
Abstract
Background: Genome-wide maps of transcription factor binding sites in primary tissues can expand our understanding of genome function, transcriptional regulation, and genetic alterations that contribute to disease risk. However, almost all genome-wide studies of transcription factors have been in cell lines, and performing these experiments in tissues has been technically challenging and limited in throughput.
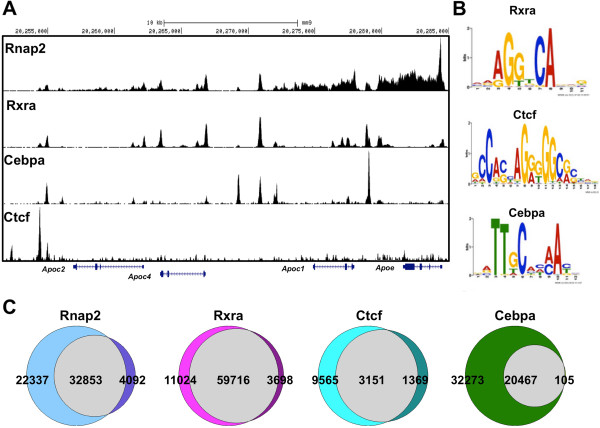
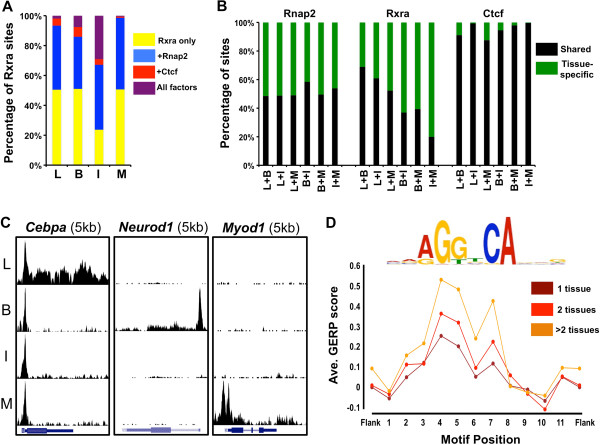
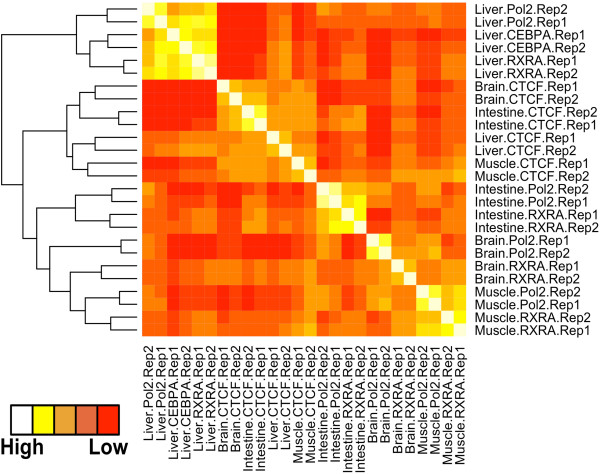
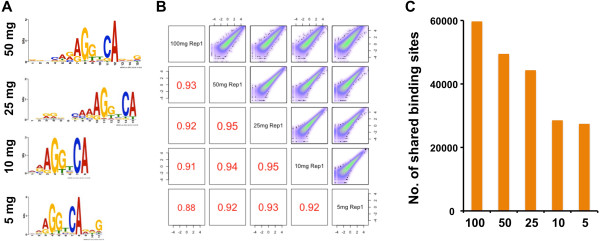
Results: Here we outline a simple strategy for mapping transcription factor binding sites in frozen tissues that utilizes dry pulverization of samples and is scalable for high-throughput analyses. We show that the method leads to accurate and reproducible chromatin immunoprecipitation next-generation sequencing (ChIP-seq) data, and is highly sensitive, identifying high-quality transcription factor binding sites from chromatin corresponding to only 5 mg of liver tissue.
Conclusions: The enhanced reproducibility, robustness, and sensitivity of the dry pulverization method, in addition to the ease of implementation and scalability, makes ChIP-seq in primary tissues a widely accessible assay.
Figures
References
-
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H. et al.Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. - DOI - PMC - PubMed
-
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, Khatun J, Lajoie BR, Landt SG, Lee B-K, Pauli F, Rosenbloom KR, Sabo P, Safi A, Sanyal A, Shoresh N, Simon JM, Song L, Trinklein ND, Altshuler RC, Birney E, Brown JB, Cheng C, Djebali S, Dong X, Ernst J. et al.An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. - DOI - PMC - PubMed
-
- Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, Absher DM, Wold BJ, Myers RM. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23(3):555–567. doi: 10.1101/gr.147942.112. - DOI - PMC - PubMed
-
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. doi: 10.1126/science.1181369. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
