

Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth
- PMID: 24280005
- PMCID: PMC3835903
- DOI: 10.1186/2049-3002-1-3
Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth
Abstract
Background: Regulation of lipid metabolism via activation of sterol regulatory element binding proteins (SREBPs) has emerged as an important function of the Akt/mTORC1 signaling axis. Although the contribution of dysregulated Akt/mTORC1 signaling to cancer has been investigated extensively and altered lipid metabolism is observed in many tumors, the exact role of SREBPs in the control of biosynthetic processes required for Akt-dependent cell growth and their contribution to tumorigenesis remains unclear.
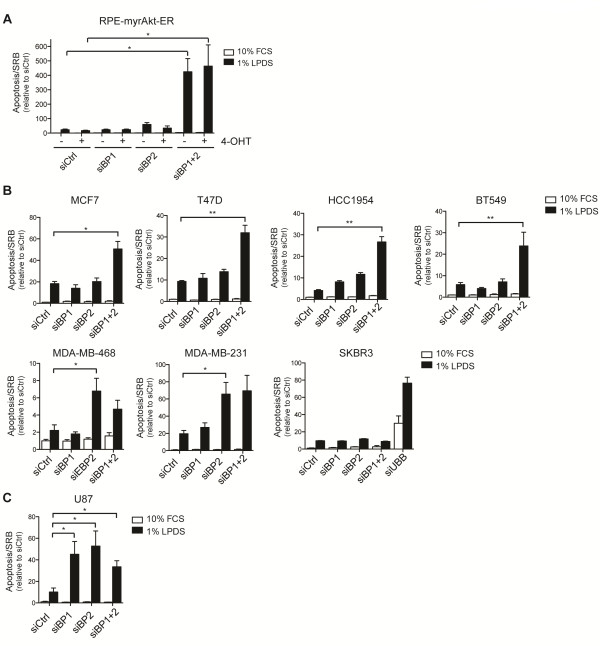
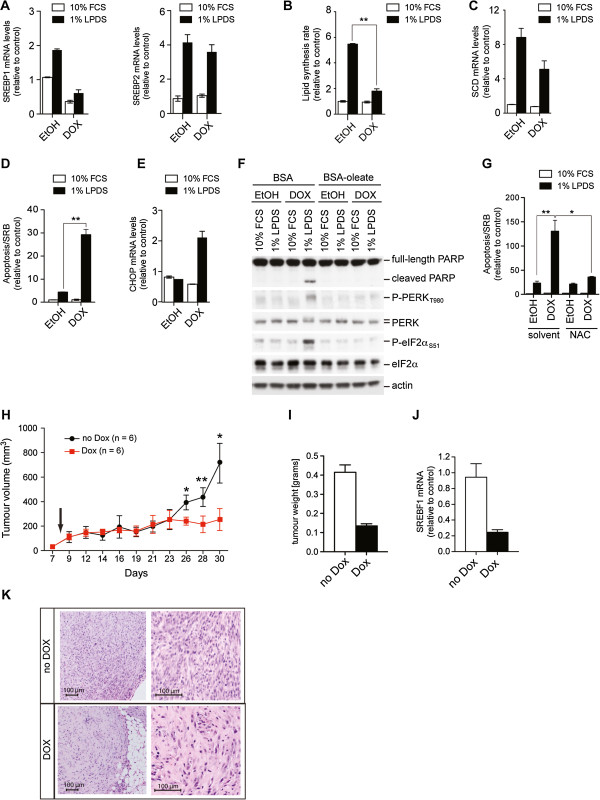
Results: We first investigated the effects of loss of SREBP function in non-transformed cells. Combined ablation of SREBP1 and SREBP2 by siRNA-mediated gene silencing or chemical inhibition of SREBP activation induced endoplasmic reticulum (ER)-stress and engaged the unfolded protein response (UPR) pathway, specifically under lipoprotein-deplete conditions in human retinal pigment epithelial cells. Induction of ER-stress led to inhibition of protein synthesis through increased phosphorylation of eIF2α. This demonstrates for the first time the importance of SREBP in the coordination of lipid and protein biosynthesis, two processes that are essential for cell growth and proliferation. SREBP ablation caused major changes in lipid composition characterized by a loss of mono- and poly-unsaturated lipids and induced accumulation of reactive oxygen species (ROS) and apoptosis. Alterations in lipid composition and increased ROS levels, rather than overall changes to lipid synthesis rate, were required for ER-stress induction.Next, we analyzed the effect of SREBP ablation in a panel of cancer cell lines. Importantly, induction of apoptosis following SREBP depletion was restricted to lipoprotein-deplete conditions. U87 glioblastoma cells were highly susceptible to silencing of either SREBP isoform, and apoptosis induced by SREBP1 depletion in these cells was rescued by antioxidants or by restoring the levels of mono-unsaturated fatty acids. Moreover, silencing of SREBP1 induced ER-stress in U87 cells in lipoprotein-deplete conditions and prevented tumor growth in a xenograft model.
Conclusions: Taken together, these results demonstrate that regulation of lipid composition by SREBP is essential to maintain the balance between protein and lipid biosynthesis downstream of Akt and to prevent resultant ER-stress and cell death. Regulation of lipid metabolism by the Akt/mTORC1 signaling axis is required for the growth and survival of cancer cells.
Figures





References
-
- Horton JD. Sterol regulatory element-binding proteins: transcriptional activators of lipid synthesis. Biochem Soc Trans. 2002;30:1091–1095. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
